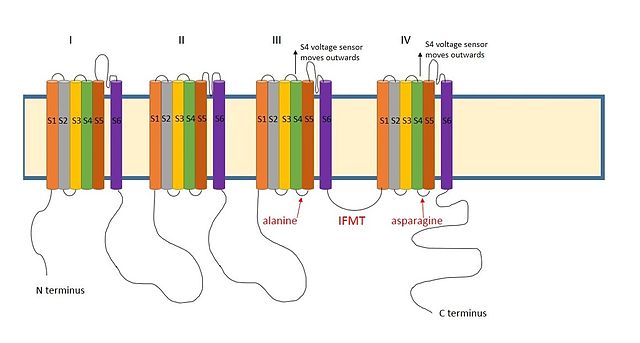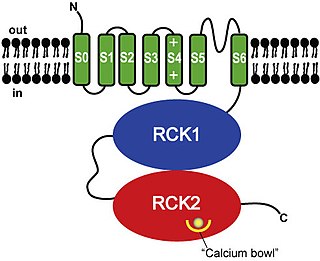
BK channels (big potassium), are large conductance calcium-activated potassium channels, also known as Maxi-K, slo1, or Kca1.1. BK channels are voltage-gated potassium channels that conduct large amounts of potassium ions (K+) across the cell membrane, hence their name, big potassium. These channels can be activated (opened) by either electrical means, or by increasing Ca2+ concentrations in the cell. BK channels help regulate physiological processes, such as circadian behavioral rhythms and neuronal excitability. BK channels are also involved in many processes in the body, as it is a ubiquitous channel. They have a tetrameric structure that is composed of a transmembrane domain, voltage sensing domain, potassium channel domain, and a cytoplasmic C-terminal domain, with many X-ray structures for reference. Their function is to repolarize the membrane potential by allowing for potassium to flow outward, in response to a depolarization or increase in calcium levels.

Potassium channels are the most widely distributed type of ion channel found in virtually all organisms. They form potassium-selective pores that span cell membranes. Potassium channels are found in most cell types and control a wide variety of cell functions.
Hyperkalemic periodic paralysis is an inherited autosomal dominant disorder that affects sodium channels in muscle cells and the ability to regulate potassium levels in the blood. It is characterized by muscle hyperexcitability or weakness which, exacerbated by potassium, heat or cold, can lead to uncontrolled shaking followed by paralysis. Onset usually occurs in early childhood, but it still occurs with adults.
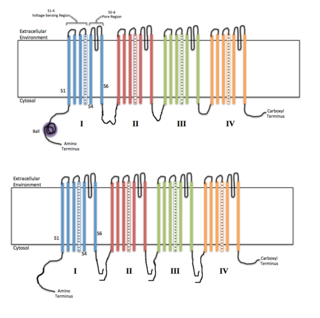
Voltage-gated ion channels are a class of transmembrane proteins that form ion channels that are activated by changes in the electrical membrane potential near the channel. The membrane potential alters the conformation of the channel proteins, regulating their opening and closing. Cell membranes are generally impermeable to ions, thus they must diffuse through the membrane through transmembrane protein channels. They have a crucial role in excitable cells such as neuronal and muscle tissues, allowing a rapid and co-ordinated depolarization in response to triggering voltage change. Found along the axon and at the synapse, voltage-gated ion channels directionally propagate electrical signals. Voltage-gated ion-channels are usually ion-specific, and channels specific to sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (Cl−) ions have been identified. The opening and closing of the channels are triggered by changing ion concentration, and hence charge gradient, between the sides of the cell membrane.
Sodium channels are integral membrane proteins that form ion channels, conducting sodium ions (Na+) through a cell's membrane. They belong to the superfamily of cation channels.

Potassium voltage-gated channel subfamily A member 1 also known as Kv1.1 is a shaker related voltage-gated potassium channel that in humans is encoded by the KCNA1 gene. Isaacs syndrome is a result of an autoimmune reaction against the Kv1.1 ion channel.
Voltage-gated potassium channels (VGKCs) are transmembrane channels specific for potassium and sensitive to voltage changes in the cell's membrane potential. During action potentials, they play a crucial role in returning the depolarized cell to a resting state.

Sodium channel protein type 4 subunit alpha is a protein that in humans is encoded by the SCN4A gene.
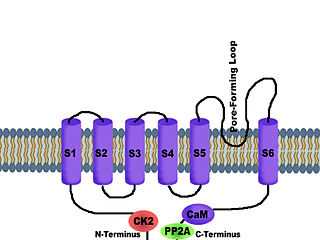
SK channels are a subfamily of calcium-activated potassium channels. They are so called because of their small single channel conductance in the order of 10 pS. SK channels are a type of ion channel allowing potassium cations to cross the cell membrane and are activated (opened) by an increase in the concentration of intracellular calcium through N-type calcium channels. Their activation limits the firing frequency of action potentials and is important for regulating afterhyperpolarization in the neurons of the central nervous system as well as many other types of electrically excitable cells. This is accomplished through the hyperpolarizing leak of positively charged potassium ions along their concentration gradient into the extracellular space. This hyperpolarization causes the membrane potential to become more negative. SK channels are thought to be involved in synaptic plasticity and therefore play important roles in learning and memory.
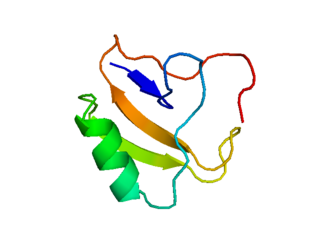
Scorpion toxins are proteins found in the venom of scorpions. Their toxic effect may be mammal- or insect-specific and acts by binding with varying degrees of specificity to members of the Voltage-gated ion channel superfamily; specifically, voltage-gated sodium channels, voltage-gated potassium channels, and Transient Receptor Potential (TRP) channels. The result of this action is to activate or inhibit the action of these channels in the nervous and cardiac organ systems. For instance, α-scorpion toxins MeuNaTxα-12 and MeuNaTxα-13 from Mesobuthus eupeus are neurotoxins that target voltage-gated Na+ channels (Navs), inhibiting fast inactivation. In vivo assays of MeuNaTxα-12 and MeuNaTxα-13 effects on mammalian and insect Navs show differential potency. These recombinants exhibit their preferential affinity for mammalian and insect Na+ channels at the α-like toxins' active site, site 3, in order to inactivate the cell membrane depolarization faster[6]. The varying sensitivity of different Navs to MeuNaTxα-12 and MeuNaTxα-13 may be dependent on the substitution of a conserved Valine residue for a Phenylalanine residue at position 1630 of the LD4:S3-S4 subunit or due to various changes in residues in the LD4:S5-S6 subunit of the Navs. Ultimately, these actions can serve the purpose of warding off predators by causing pain or to subdue predators.
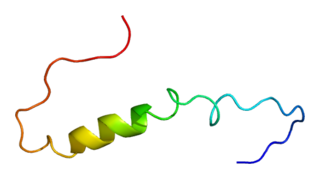
Calcium-activated potassium channel subunit beta-2 is a protein that in humans is encoded by the KCNMB2 gene.
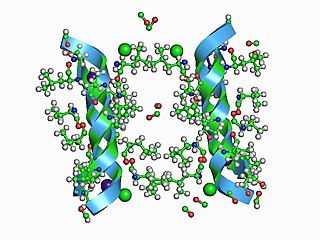
In electrophysiology, the term gating refers to the opening (activation) or closing of ion channels. This change in conformation is a response to changes in transmembrane voltage.
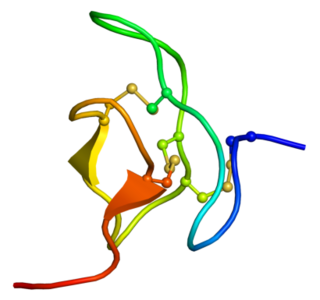
Phrixotoxins are peptide toxins derived from the venom of the Chilean copper tarantula Phrixotrichus auratus, also named Paraphysa scrofa. Phrixotoxin-1 and -2 block A-type voltage-gated potassium channels; phrixotoxin-3 blocks voltage-gated sodium channels. Similar toxins are found in other species, for instance the Chilean rose tarantula.

Heteroscodratoxin-1 is a neurotoxin produced by the venom glands of Heteroscodra maculata that shifts the activation threshold of voltage-gated potassium channels and the inactivation of Nav1.1 sodium channels to more positive potentials.

KcsA (Kchannel of streptomyces A) is a prokaryotic potassium channel from the soil bacterium Streptomyces lividans that has been studied extensively in ion channel research. The pH activated protein possesses two transmembrane segments and a highly selective pore region, responsible for the gating and shuttling of K+ ions out of the cell. The amino acid sequence found in the selectivity filter of KcsA is highly conserved among both prokaryotic and eukaryotic K+ voltage channels; as a result, research on KcsA has provided important structural and mechanistic insight on the molecular basis for K+ ion selection and conduction. As one of the most studied ion channels to this day, KcsA is a template for research on K+ channel function and its elucidated structure underlies computational modeling of channel dynamics for both prokaryotic and eukaryotic species.
Halcurin is a polypeptide neurotoxin from the sea anemone Halcurias sp. Based on sequence homology to type 1 and type 2 sea anemone toxins it is thought to delay channel inactivation by binding to the extracellular site 3 on the voltage gated sodium channels in a membrane potential-dependent manner.
Blood-depressing substance-1 (BDS-1), also known as kappa-actitoxin-Avd4a, is a polypeptide found in the venom of the snakelocks anemone Anemonia sulcata. BDS-1 is a neurotoxin that modulates voltage-dependent potassium channels, in particular Kv3-family channels, as well as certain sodium channels. This polypeptide belongs to the sea anemone type 3 toxin peptide family.
HsTx1 is a toxin from the venom of the scorpion Heterometrus spinifer. HsTx1 is a very potent inhibitor of the rat Kv1.3 voltage-gated potassium channel.
LmαTX3 is an α-scorpion toxin from Lychas mucronatus. that inhibits fast inactivation of voltage gated sodium-channels (VGSCs).
Protoxin-I, also known as ProTx-I, or Beta/omega-theraphotoxin-Tp1a, is a 35-amino-acid peptide neurotoxin extracted from the venom of the tarantula Thrixopelma pruriens. Protoxin-I belongs to the inhibitory cystine knot (ICK) family of peptide toxins, which have been known to potently inhibit voltage-gated ion channels. Protoxin-I selectively blocks low voltage threshold T-type calcium channels, voltage gated sodium channels and the nociceptor cation channel TRPA1. Due to its unique ability to bind to TRPA1, Protoxin-I has been implicated as a valuable pharmacological reagent with potential applications in clinical contexts with regards to pain and inflammation