Related Research Articles

Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
Rett syndrome (RTT) is a genetic disorder that typically becomes apparent after 6–18 months of age and almost exclusively in females. Symptoms include impairments in language and coordination, and repetitive movements. Those affected often have slower growth, difficulty walking, and a smaller head size. Complications of Rett syndrome can include seizures, scoliosis, and sleeping problems. The severity of the condition is variable.

Ehlers–Danlos syndromes (EDS) are a group of 14 genetic connective-tissue disorders in the current classification, with the latest type discovered in 2018. Symptoms include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.
Advanced Sleep Phase Disorder (ASPD), also known as the advanced sleep-phase type (ASPT) of circadian rhythm sleep disorder, is a condition that is characterized by a recurrent pattern of early evening sleepiness and very early morning awakening. This sleep phase advancement can interfere with daily social and work schedules, and results in shortened sleep duration and excessive daytime sleepiness. The timing of sleep and melatonin levels are regulated by the body's central circadian clock, which is located in the suprachiasmatic nucleus in the hypothalamus.

Long QT syndrome (LQTS) is a condition affecting repolarization (relaxing) of the heart after a heartbeat, giving rise to an abnormally lengthy QT interval. It results in an increased risk of an irregular heartbeat which can result in fainting, drowning, seizures, or sudden death. These episodes can be triggered by exercise or stress. Some rare forms of LQTS are associated with other symptoms and signs including deafness and periods of muscle weakness.

Occipital horn syndrome (OHS), formerly considered a variant of Ehlers–Danlos syndrome, is an X-linked recessive mitochondrial and connective tissue disorder. It is caused by a deficiency in the transport of the essential mineral copper, associated with mutations in the ATP7A gene.
Timothy syndrome is a rare autosomal-dominant disorder characterized by physical malformations, as well as neurological and developmental defects, including heart QT-prolongation, heart arrhythmias, structural heart defects, syndactyly, and autism spectrum disorders. Timothy syndrome represents one clinical manifestation of a range of disorders associated with mutations in CACNA1C, the gene encoding the calcium channel Cav1.2 α subunit.
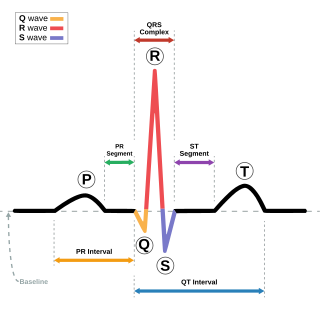
Romano–Ward syndrome is the most common form of congenital Long QT syndrome (LQTS), a genetic heart condition that affects the electrical properties of heart muscle cells. Those affected are at risk of abnormal heart rhythms which can lead to fainting, seizures, or sudden death. Romano–Ward syndrome can be distinguished clinically from other forms of inherited LQTS as it affects only the electrical properties of the heart, while other forms of LQTS can also affect other parts of the body.
Voltage-gated calcium channels (VGCCs), also known as voltage-dependent calcium channels (VDCCs), are a group of voltage-gated ion channels found in the membrane of excitable cells (e.g., muscle, glial cells, neurons, etc.) with a permeability to the calcium ion Ca2+. These channels are slightly permeable to sodium ions, so they are also called Ca2+-Na+ channels, but their permeability to calcium is about 1000-fold greater than to sodium under normal physiological conditions.

Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.
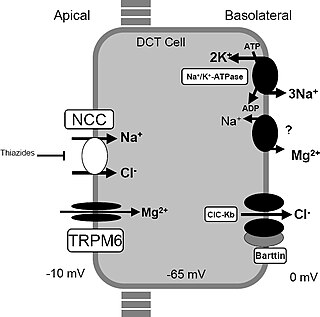
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. The disorder is caused by disease-causing variants in both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney. The distal convoluted tubule of the kidney plays an important homeostatic role in sodium and chloride absorption as well as of the reabsorption of magnesium and calcium.

The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis, although the genetics of autism are complex and it is unclear whether autism spectrum disorder (ASD) is explained more by multigene interactions or by rare mutations with major effects.
Farber disease is an extremely rare, progressive, autosomal recessive lysosomal storage disease caused by a deficiency of the acid ceramidase enzyme. Acid ceramidase is responsible for breaking down ceramide into sphingosine and fatty acid. When the enzyme is deficient, this leads to an accumulation of fatty material in the lysosomes of the cells, leading to the signs and symptoms of this disorder.

McLeod syndrome is an X-linked recessive genetic disorder that may affect the blood, brain, peripheral nerves, muscle, and heart. It is caused by a variety of recessively inherited mutations in the XK gene on the X chromosome. The gene is responsible for producing the Kx protein, a secondary supportive protein for the Kell antigen on the red blood cell surface.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Calcium channel, voltage-dependent, L type, alpha 1C subunit is a protein that in humans is encoded by the CACNA1C gene. Cav1.2 is a subunit of L-type voltage-dependent calcium channel.

Cav2.1, also called the P/Q voltage-dependent calcium channel, is a calcium channel found mainly in the brain. Specifically, it is found on the presynaptic terminals of neurons in the brain and cerebellum. Cav2.1 plays an important role in controlling the release of neurotransmitters between neurons. It is composed of multiple subunits, including alpha-1, beta, alpha-2/delta, and gamma subunits. The alpha-1 subunit is the pore-forming subunit, meaning that the calcium ions flow through it. Different kinds of calcium channels have different isoforms (versions) of the alpha-1 subunit. Cav2.1 has the alpha-1A subunit, which is encoded by the CACNA1A gene. Mutations in CACNA1A have been associated with various neurologic disorders, including familial hemiplegic migraine, episodic ataxia type 2, and spinocerebellar ataxia type 6.
Mitochondrially encoded tRNA histidine, also known as MT-TH, is a transfer RNA which, in humans, is encoded by the mitochondrial MT-TH gene.
Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.
L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.
References
- 1 2 Levy, Rebecca J.; Timothy, Katherine W.; Underwood, Jack F.G.; Hall, Jeremy; Bernstein, Jonathan A.; Pașca, Sergiu P. (January 2023). "A Cross-Sectional Study of the Neuropsychiatric Phenotype of CACNA1C-Related Disorder". Pediatric Neurology. 138: 101–106. doi:10.1016/j.pediatrneurol.2022.10.013. PMID 36436328.
- 1 2 Napolitano, Carlo; Timothy, Katherine W.; Bloise, Raffaella; Priori, Silvia G. (1993), Adam, Margaret P.; Everman, David B.; Mirzaa, Ghayda M.; Pagon, Roberta A. (eds.), "CACNA1C-Related Disorders", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301577 , retrieved 2022-12-12
- ↑ Rodan, Lance H.; Spillmann, Rebecca C.; Kurata, Harley T.; Lamothe, Shawn M.; Maghera, Jasmine; Jamra, Rami Abou; Alkelai, Anna; Antonarakis, Stylianos E.; Atallah, Isis; Bar-Yosef, Omer; Bilan, Frédéric; Bjorgo, Kathrine; Blanc, Xavier; Van Bogaert, Patrick; Bolkier, Yoav (October 2021). "Phenotypic expansion of CACNA1C-associated disorders to include isolated neurological manifestations". Genetics in Medicine. 23 (10): 1922–1932. doi:10.1038/s41436-021-01232-8. PMC 8488020 . PMID 34163037.
- ↑ Bauer, Rosemary; Timothy, Katherine W.; Golden, Andy (2021-05-17). "Update on the Molecular Genetics of Timothy Syndrome". Frontiers in Pediatrics. 9: 668546. doi: 10.3389/fped.2021.668546 . ISSN 2296-2360. PMC 8165229 . PMID 34079780.
| | This genetics article is a stub. You can help Wikipedia by expanding it. |