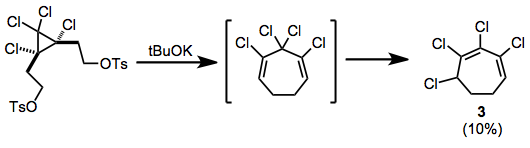
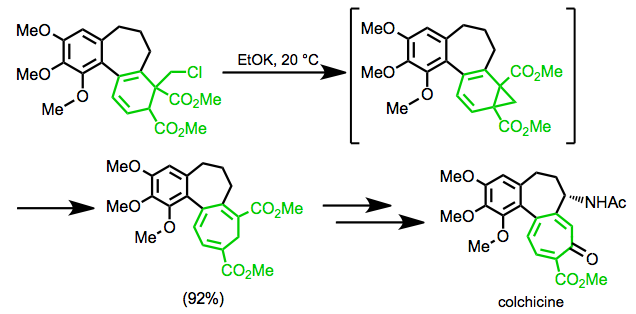
In organic chemistry, an electrocyclic reaction is a type of pericyclic rearrangement where the net result is one pi bond being converted into one sigma bond or vice versa. These reactions are usually categorized by the following criteria:
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
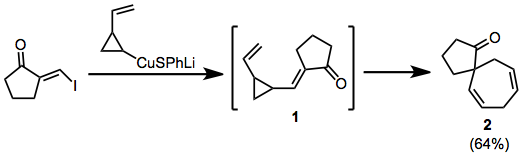
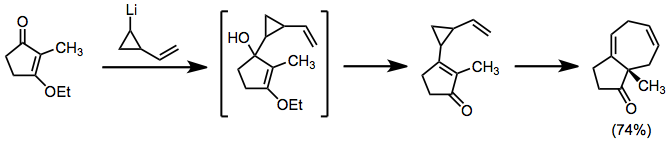
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
The Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, in organic chemistry is a special type of elimination reaction in which two vicinal substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents unlike regular eliminations which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
The α-ketol rearrangement is the acid-, base-, or heat-induced 1,2-migration of an alkyl or aryl group in an α-hydroxy ketone or aldehyde to give an isomeric product.
Organostannane addition reactions comprise the nucleophilic addition of an allyl-, allenyl-, or propargylstannane to an aldehyde, imine, or, in rare cases, a ketone. The reaction is widely used for carbonyl allylation.
Vicinal difunctionalization refers to a chemical reaction involving transformations at two adjacent centers. This transformation can be accomplished in α,β-unsaturated carbonyl compounds via the conjugate addition of a nucleophile to the β-position followed by trapping of the resulting enolate with an electrophile at the α-position. When the nucleophile is an enolate and the electrophile a proton, the reaction is called Michael addition.
Electrophilic substitution of unsaturated silanes involves attack of an electrophile on an allyl- or vinylsilane. An allyl or vinyl group is incorporated at the electrophilic center after loss of the silyl group.
Ketene cycloadditions are the reactions of the pi system of ketenes with unsaturated compounds to provide four-membered or larger rings. [2+2], [3+2], and [4+2] variants of the reaction are known.
Carbonyl oxidation with hypervalent iodine reagents involves the functionalization of the α position of carbonyl compounds through the intermediacy of a hypervalent iodine(III) enolate species. This electrophilic intermediate may be attacked by a variety of nucleophiles or undergo rearrangement or elimination.
Oxidation with chromium(VI) complexes involves the conversion of alcohols to carbonyl compounds or more highly oxidized products through the action of molecular chromium(VI) oxides and salts. The principal reagents are Collins reagent, PDC, and PCC. These reagents represent improvements over inorganic chromium(VI) reagents such as Jones reagent.
The Payne rearrangement is the isomerization, under basic conditions, of 2,3-epoxy alcohols to isomeric 1,2-epoxy alcohols with inversion of configuration. Aza- and thia-Payne rearrangements of aziridines and thiiraniums, respectively, are also known.
Reductions with metal alkoxyaluminium hydrides are chemical reactions that involve either the net hydrogenation of an unsaturated compound or the replacement of a reducible functional group with hydrogen by metal alkoxyaluminium hydride reagents.
Metal-catalyzed intermolecular carbenoid cyclopropanations are organic reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc.
Manganese-mediated coupling reactions are radical coupling reactions between enolizable carbonyl compounds and unsaturated compounds initiated by a manganese(III) salt, typically manganese(III) acetate. Copper(II) acetate is sometimes used as a co-oxidant to assist in the oxidation of intermediate radicals to carbocations.
Reactions of alkenyl- and alkynylaluminium compounds involve the transfer of a nucleophilic alkenyl or alkynyl group attached to aluminium to an electrophilic atom. Stereospecific hydroalumination, carboalumination, and terminal alkyne metalation are useful methods for generation of the necessary alkenyl- and alkynylalanes.
In organic chemistry, the oxy-Cope rearrangement is a chemical reaction. It involves reorganization of the skeleton of certain unsaturated alcohols. It is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.