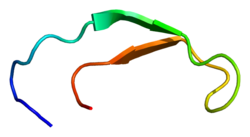During conditions in which the hepcidin level is abnormally high, such as inflammation, serum iron falls due to iron trapping within macrophages and liver cells and decreased gut iron absorption. This typically leads to anemia due to an inadequate amount of blood serum iron being available for developing red blood cells. When the hepcidin level is abnormally low, such as in hemochromatosis, iron overload occurs due to increased ferroportin mediated iron efflux from storage and increased gut iron absorption.
Structure
Hepcidin is initially synthesized as an 84-amino acid preprohormone (preprohepcidin) which undergoes sequential cleavages to form the active, mature hormone. The first cleavage by signal peptidase removes the 24-amino acid N-terminal signal peptide, creating a 60-amino acid prohepcidin. Furin-like convertase[7] and α-1 antitrypsin[8] then cleave prohepcidin to remove a 35-amino acid proregion, resulting in the 25-amino acid mature, bioactive hepcidin. There are also shorter isoforms of hepcidin, with 20 and 22 amino acids, which have minimal iron regulatory activity. Only the 9 N-terminal amino acids are essential for hepcidin's biological activity, specifically its ability to bind to ferroportin and regulate iron metabolism.
Structurally, hepcidin is a tightly folded polypeptide with 32% beta-sheet character and a hairpin tertiary structure stabilized by four disulfide bonds among eight cystein residues (crucial structure). Hepcidin's structure has been elucidated through solution NMR,[9] revealing that it interconverts between two conformations at different temperatures. X-ray analysis of a co-crystal with Fab confirmed a structure similar to the high-temperature NMR structure.[10]
Diagram showing how hepcidin controls ferroportin levels, which in turn control entry of iron into the circulation
Hepcidin is a regulator of iron metabolism. It inhibits iron transport by binding to the iron export channel ferroportin which is located in the basolateral plasma membrane of gut enterocytes and the plasma membrane of reticuloendothelial cells (macrophages), ultimately resulting in ferroportin breakdown in lysosomes.[11][12] It has been shown that hepcidin is able to bind to the central cavity of ferroportin, thus occluding iron export from the cell. This suggests that hepcidin is able to regulate iron export independently of ferroportin endocytosis and ubiquitination, and is thus quickly inducible and reversible.[13][14]
In enterocytes, this prevents iron transmission into the hepatic portal system, thereby reducing dietary iron absorption. In macrophages, ferroportin inhibition causes iron be to stored within the cell. Increased hepcidin activity is partially responsible for reduced iron availability seen in anemia of chronic inflammation, such as kidney failure; this may explain why patients with end stage kidney failure may not respond to oral iron replacement.[15]
Any one of several mutations in hepcidin will result in juvenile hemochromatosis. The majority of juvenile hemochromatosis cases are caused by mutations in hemojuvelin.[16] Mutations in TMPRSS6 can cause anemia through dysregulation of hepcidin.[17]
Hepcidin creation (synthesis) and secretion by the liver is controlled by iron stores, inflammation (hepcidin is an acute phase reactant), hypoxia, and production of red blood cells (erythropoiesis).[19] In response to large iron stores, production of bone morphogenic protein (BMP) is induced, which binds to receptors on hepatocytes and induces hepcidin expression via the SMAD pathway.[20] Inflammation causes an increase in hepcidin production by releasing the signaling molecule interleukin-6 (IL-6), which binds to a receptor and upregulates the HAMP gene via the JAK/STAT pathway.[20] Hypoxia negatively regulates hepcidin production via production the transcription factor hypoxia-inducible factor (HIF), which under normal conditions is degraded by von Hippel-Lindau (VHL) and prolyl dehydrogenase (PHD). However, when hypoxia is induced, PHD is inactivated, thus allowing HIF to down-regulate hepcidin production. Erythropoiesis decreases hepcidin production via production of erythropoietin (EPO), which has been shown to down-regulate hepcidin production.[20]
Severe anemia is associated with low hepcidin levels, even in the presence of inflammation.[21]Erythroferrone, produced in red blood cells (erythroblasts), has been identified as inhibiting hepcidin, thus providing more iron for hemoglobin synthesis in situations such as stress erythropoiesis.[22][23]
Vitamin D has been shown to decrease hepcidin, both in cell models looking at transcription and when given in large doses to human volunteers. Optimal function of hepcidin may require adequate levels of vitamin D in the blood.[24]
History
Hepcidin was initially reported and named in January 1998,[18] after it was observed that it was produced in the liver and appeared to have bactericidal (bacteria-killing) properties. Detailed descriptions were published in 2000–2001.[25][26][27] Although it is primarily synthesized in the liver, smaller amounts are synthesized in other tissues such as fat cells.[28]
Hepcidin was first discovered in humanurine and blood serum.[29] Soon after this discovery, researchers discovered that hepcidin production in mice increases in conditions of iron overload as well as inflammation. Genetically modified mice engineered to overexpress hepcidin died shortly after birth with severe iron deficiency, again suggesting that hepcidin plays a central and not redundant role in iron regulation.
The first piece of evidence that linked hepcidin to the clinical condition known as the anemia of inflammation came from the lab of Nancy Andrews in Boston, when researchers looked at tissue from two patients with liver tumors with a severe microcytic anemia that did not respond to iron supplements. The tumor tissue appeared to be overproducing hepcidin, and contained large quantities of hepcidin mRNA. Removing the tumors surgically cured the anemia.[citation needed]
Taken together, these discoveries suggested that hepcidin regulates the absorption of iron into the body.
Hepcidin (blue) bound to the central cavity of ferroportin
Clinical significance
There are many diseases where failure to adequately absorb iron contributes to iron deficiency and iron deficiency anemia. The treatment will depend on the hepcidin levels that are present, as oral treatment will be unlikely to be effective if hepcidin is blocking enteral absorption; in these cases, parenteral iron treatment would be appropriate. Studies have found that measuring hepcidin would help establish the optimal treatment for a patient,[30] but as this is not widely available, C-reactive protein (CRP) is used as a surrogate marker.
Beta thalassemia, one of the most common congenitalanemias, arises from partial or complete failure to synthesize beta-globin, a component of hemoglobin. Excessive iron absorption is one of the main features of beta thalassemia and can lead to severe morbidity and mortality. The serial analyses of beta thalassemic mice indicate that hemoglobin levels decrease over time, while the concentration of iron in the liver, spleen, and kidneys increases significantly. The overload of iron is associated with low levels of hepcidin. Patients with beta thalassemia also have low hepcidin levels. The observations led researchers to hypothesize that more iron is absorbed in beta thalassemia than is required for erythropoiesis. Increasing expression of hepcidin in beta thalassemic mice limits iron overload, and also decreases formation of insoluble membrane-bound globins and reactive oxygen species, and improves anemia.[32] Mice with increased hepcidin expression also demonstrated an increase in the lifespan of their red cells, reversal of ineffective erythropoiesis and splenomegaly, and an increase in total hemoglobin levels. From these data, researchers suggested that therapeutics to increase hepcidin levels or act as hepcidin agonists could help treat the abnormal iron absorption in individuals with beta thalassemia and related disorders.[33] In later studies with mice,[34]erythroferrone has been suggested to be the factor that is responsible for the hepcidin suppression. Correcting hepcidin and iron levels in these mice did not improve their anemia.[34]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.