Related Research Articles

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.
The International HapMap Project was an organization that aimed to develop a haplotype map (HapMap) of the human genome, to describe the common patterns of human genetic variation. HapMap is used to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available for research.

A DNA segment is identical by state (IBS) in two or more individuals if they have identical nucleotide sequences in this segment. An IBS segment is identical by descent (IBD) in two or more individuals if they have inherited it from a common ancestor without recombination, that is, the segment has the same ancestral origin in these individuals. DNA segments that are IBD are IBS per definition, but segments that are not IBD can still be IBS due to the same mutations in different individuals or recombinations that do not alter the segment.

Human genetic variation is the genetic differences in and among populations. There may be multiple variants of any given gene in the human population (alleles), a situation called polymorphism.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.

The 1000 Genomes Project (1KGP), taken place from January 2008 to 2015, was an international research effort to establish the most detailed catalogue of human genetic variation at the time. Scientists planned to sequence the genomes of at least one thousand anonymous healthy participants from a number of different ethnic groups within the following three years, using advancements in newly developed technologies. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.

The Single Nucleotide Polymorphism Database (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI). Although the name of the database implies a collection of one class of polymorphisms only, it in fact contains a range of molecular variation: (1) SNPs, (2) short deletion and insertion polymorphisms (indels/DIPs), (3) microsatellite markers or short tandem repeats (STRs), (4) multinucleotide polymorphisms (MNPs), (5) heterozygous sequences, and (6) named variants. The dbSNP accepts apparently neutral polymorphisms, polymorphisms corresponding to known phenotypes, and regions of no variation. It was created in September 1998 to supplement GenBank, NCBI’s collection of publicly available nucleic acid and protein sequences.
Whole genome sequencing (WGS), also known as full genome sequencing, complete genome sequencing, or entire genome sequencing, is the process of determining the entirety, or nearly the entirety, of the DNA sequence of an organism's genome at a single time. This entails sequencing all of an organism's chromosomal DNA as well as DNA contained in the mitochondria and, for plants, in the chloroplast.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.
In genetics, haplotype estimation refers to the process of statistical estimation of haplotypes from genotype data. The most common situation arises when genotypes are collected at a set of polymorphic sites from a group of individuals. For example in human genetics, genome-wide association studies collect genotypes in thousands of individuals at between 200,000-5,000,000 SNPs using microarrays. Haplotype estimation methods are used in the analysis of these datasets and allow genotype imputation of alleles from reference databases such as the HapMap Project and the 1000 Genomes Project.
Project MinE is an independent large scale whole genome research project that was initiated by 2 patients with amyotrophic lateral sclerosis and started on World ALS Day, June 21, 2013.
SNV calling from NGS data is any of a range of methods for identifying the existence of single nucleotide variants (SNVs) from the results of next generation sequencing (NGS) experiments. These are computational techniques, and are in contrast to special experimental methods based on known population-wide single nucleotide polymorphisms. Due to the increasing abundance of NGS data, these techniques are becoming increasingly popular for performing SNP genotyping, with a wide variety of algorithms designed for specific experimental designs and applications. In addition to the usual application domain of SNP genotyping, these techniques have been successfully adapted to identify rare SNPs within a population, as well as detecting somatic SNVs within an individual using multiple tissue samples.
Mega2 allows the applied statistical geneticist to convert one's data from several input formats to a large number output formats suitable for analysis by commonly used software packages. In a typical human genetics study, the analyst often needs to use a variety of different software programs to analyze the data, and these programs usually require that the data be formatted to their precise input specifications. Conversion of one's data into these multiple different formats can be tedious, time-consuming, and error-prone. Mega2, by providing validated conversion pipelines, can accelerate the analyses while reducing errors.
Karen L. Mohlke is a biologist at University of North Carolina, Chapel Hill. She is known for her work in human genetics, especially in the area of diabetes research. She was one of the first researchers to use exome array genotyping.
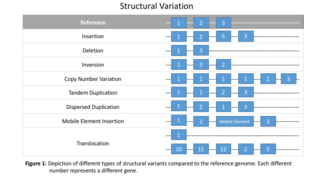
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.

Jonathan Laurence Marchini is a Bayesian statistician and professor of statistical genomics in the Department of Statistics at the University of Oxford, a tutorial fellow in statistics at Somerville College, Oxford and a co-founder and director of Gensci Ltd. He co-leads the Haplotype Reference Consortium.

The Tohoku Medical Megabank Project is a national project in Japan. The mission of the Tohoku Medical Megabank (TMM) project is to carry out a long-term health survey in the Miyagi and Iwate prefectures, which were affected by the Great East Japan Earthquake, and provide the research infrastructure for the development of personalized medicine by establishing a biobank and conducting cohort studies. It started in 2012.
Personalized genomics is the human genetics-derived study of analyzing and interpreting individualized genetic information by genome sequencing to identify genetic variations compared to the library of known sequences. International genetics communities have spared no effort from the past and have gradually cooperated to prosecute research projects to determine DNA sequences of the human genome using DNA sequencing techniques. The methods that are the most commonly used are whole exome sequencing and whole genome sequencing. Both approaches are used to identify genetic variations. Genome sequencing became more cost-effective over time, and made it applicable in the medical field, allowing scientists to understand which genes are attributed to specific diseases.
References
- 1 2 Scheet, Paul; Stephens, Matthew (2006). "A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase". The American Journal of Human Genetics. 78 (4): 629–644. doi:10.1086/502802. PMC 1424677 . PMID 16532393.
- ↑ Marchini, J.; Howie, B. (2010). "Genotype imputation for genome-wide association studies". Nature Reviews Genetics. 11 (7): 499–511. doi:10.1038/nrg2796. PMID 20517342. S2CID 1465707.
- 1 2 Deng, T; Zhang, P; Garrick, D; Gao, H; Wang, L; Zhao, F (2021). "Comparison of Genotype Imputation for SNP Array and Low-Coverage Whole-Genome Sequencing Data". Frontiers in Genetics. 12: 704118. doi: 10.3389/fgene.2021.704118 . PMC 8762119 . PMID 35046990.
- ↑ Sousa da Mota, Bárbara; Rubinacci, Simone; Cruz Dávalos, Diana Ivette; G. Amorim, Carlos Eduardo; Sikora, Martin; Johannsen, Niels N.; Szmyt, Marzena H.; Włodarczak, Piotr; Szczepanek, Anita; Przybyła, Marcin M.; Schroeder, Hannes; Allentoft, Morten E.; Willerslev, Eske; Malaspinas, Anna-Sapfo; Delaneau, Olivier (20 June 2023). "Imputation of ancient human genomes". Nature Communications. 14 (1): 3660. Bibcode:2023NatCo..14.3660S. doi:10.1038/s41467-023-39202-0. PMC 10282092 . PMID 37339987.
- ↑ Li, Y; Willer, CJ; Ding, J; Scheet, P; Abecasis, GR (Dec 2010). "MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes". Genetic Epidemiology. 34 (8): 816–34. doi:10.1002/gepi.20533. PMC 3175618 . PMID 21058334.
- ↑ Howie, B; Fuchsberger, C; Stephens, M; Marchini, J; Abecasis, GR (Jul 22, 2012). "Fast and accurate genotype imputation in genome-wide association studies through pre-phasing". Nature Genetics. 44 (8): 955–9. doi:10.1038/ng.2354. PMC 3696580 . PMID 22820512.
- ↑ Browning, Brian L.; Browning, Sharon R. (2009). "A Unified Approach to Genotype Imputation and Haplotype-Phase Inference for Large Data Sets of Trios and Unrelated Individuals". The American Journal of Human Genetics. 84 (2): 210–223. doi:10.1016/j.ajhg.2009.01.005. PMC 2668004 . PMID 19200528.
- ↑ Howie, Bryan; Fuchsberger, Christian; Stephens, Matthew; Marchini, Jonathan; Abecasis, Gonçalo R (22 July 2012). "Fast and accurate genotype imputation in genome-wide association studies through pre-phasing". Nature Genetics. 44 (8): 955–959. doi:10.1038/ng.2354. PMC 3696580 . PMID 22820512.
- ↑ Delaneau, Olivier; Marchini, Jonathan; Zagury, Jean-François (4 December 2011). "A linear complexity phasing method for thousands of genomes". Nature Methods. 9 (2): 179–181. doi:10.1038/nmeth.1785. PMID 22138821. S2CID 13765612.
- ↑ Durbin, Richard M.; Altshuler, David L.; Durbin, Richard M.; Abecasis, Gonçalo R.; Bentley, David R.; Chakravarti, Aravinda; Clark, Andrew G.; Collins, Francis S. (28 October 2010). "A map of human genome variation from population-scale sequencing". Nature. 467 (7319): 1061–1073. Bibcode:2010Natur.467.1061T. doi:10.1038/nature09534. PMC 3042601 . PMID 20981092.
- ↑ "1000 Genomes - A Deep Catalog of Human Genetic Variation" . Retrieved 17 July 2014.
- ↑ Howie, Bryan; Donnelly, Peter; Marchini, Jonathan (2009). "A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies". PLOS Genetics. 5 (6): e1000529. doi: 10.1371/journal.pgen.1000529 . PMC 2689936 . PMID 19543373.
- ↑ De Marino, A; Mahmoud, AA; Bose, M; Bircan, KO; Terpolovsky, A; Bamunusinghe, V; Bohn, S; Khan, U; Novković, B; Yazdi, PG (2022). "A comparative analysis of current phasing and imputation software". PLOS ONE. 17 (10): e0260177. Bibcode:2022PLoSO..1760177D. doi: 10.1371/journal.pone.0260177 . PMC 9581364 . PMID 36260643.