Malonyl-CoA decarboxylase (EC4.1.1.9), (which can also be called MCD and malonyl-CoA carboxyl-lyase) is found in bacteria and humans and has important roles in regulating fatty acid metabolism and food intake, and it is an attractive target for drug discovery. It is an enzyme associated with Malonyl-CoA decarboxylase deficiency. In humans, it is encoded by the MLYCD gene.
MCD presents two isoforms which can be transcribed form one gene: a long isoform (54kDa), distributed in mitochondria, and a short isoform (49kDa) that can be found in peroxisomes and cytosol. The long isoform includes a sequence of signaling towards mitochondria in the N-terminus; whereas the short one only contains the typical sequence of peroxisomal signaling PTS1 in the C-terminus, also shared by the long isoform.
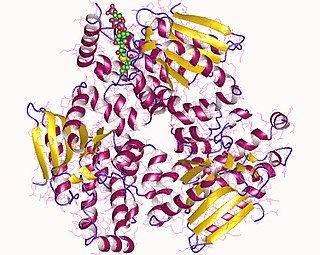
MCD is a protein tetramer, an oligomer formed by a dimer of heterodimers related by an axis of binary symmetry with a rotation angle of about 180 degrees. The strong structural asymmetry between the monomers of the heterodimer suggests a half of the sites reactivity, in which only half of the active sites are functional simultaneously. Each monomer contains basically two domains:
The N-terminus one, which is involved in oligomerization and has a helical structure of eight helixes organised as a bundle of four antiparallel helixes with two pairs of inserted helixes.
The C-terminus one is where malonyl-CoA catalysis takes place and which is present in GCN5- Histone acetyiltranferase family. It also includes a cluster of seven helixes.
However, the binding site for malonyl-CoA in MCD presents a variation with respect to their homologous: the center of the binding site has a glutamic residue instead of a glycine, acting as a molecular lever in the substrate releasing.
As said before, MCD presents a half of the sites reactivity, due to the fact that each heterodimer has two different structural conformations: B state (bound), in which the substrate is united; and U conformation (unbound), where the substrate union isn't allowed. According to this, the half of the sites mechanism might present a consumption of catalytic energy. Nevertheless, the conformational change produced in a subunit when changing from the B state to the U state (which produces the release of the product) coincides with the formation of a new union site in the active site of the neighbour subunit when changing from the U stat to B state. As a result, the conformational changes synchronised in the pair of subunits facilitates the catalysis despite the reduction of the number of available active sites.
Each monomer of that structure exhibits a large hydrophobic interface with the possibility to form an inter subunit disulfide bridge. Heterodimers are also interconnected by a small C-terminus domain interface, where a pair of cysteines is properly disposed. The disulfide bonds gives to MCD the capability to form a tetrameric enzyme linked by inter subunits covalent bonds in the presence of oxidants such as hydrogen peroxide.
MLYCD is strongly expressed in heart, liver and some other tissues like kidney. This gene is also weakly expressed in many other tissues such as brain, placenta, testis, etc.[3][4][5]
Processing and post-translational modifications
Malonyl-CoA decarboxylase is firstly processed as a pro-protein or proenzyme, in which the transit peptide, whose role is to transport the enzyme to a specific organelle (in this case the mitochondria), comprises the first 39 amino acids (beginning with a methionine and ending with an alanine). The polypeptide chain in the mature protein is comprised between amino acid 40 and 493.
In order to turn into an active enzyme, MCD undergoes 8 post-translational modifications (PTM) in different amino acids. The last one, which consists of an acetylation in the amino acid lysine in position 472, activates malonyl-CoA decarboxylase activity. Similarly, a deacetylation in this specific amino acid by SIRT4 (a mitochondrial protein) represses the enzyme activity, inhibiting fatty acid oxidation in muscle cells.[6] Another important PTM is the formation of an interchain disulfide bond in the amino acid cysteine in position 206, which may take place in peroxisomes, as the cytosolic and mitochondrial environments are too reducing for this process.[3]
Functions
The enzyme malonyl-CoA decarboxylase (MCD) functions as an indirect via of conversion from malonic semi aldehyde to acetyl-CoA in peroxisomes. This is due to the fact that the beta oxidation of long chain fatty acids with an odd number of carbons produces propionyl-CoA. Most part of this metabolite is transformed into succinyl-CoA, which is an intermediate of the tricarboxylic acid cycle. The major alternative route by which the propionyl-CoA is metabolized is based on its conversion to acrylyl-CoA. After that, it is converted to 3-hydroxy propionic acid and finally to malonic semi-aldehyde. As soon as malonic semi aldehyde is produced, it is indirectly transformed into acetyl-CoA. This conversion has been detected only in bacteria,[7] in the other natural kingdoms there is no scientific evidence to prove it.[8]
Malonyl-CoA is an important metabolite in some parts of the cell. In peroxisomes, the accumulation of this substance causes malonic aciduria, a highly pathogenic disease. To avoid it malonyl-CoA decarboxylase (MCD) converts malonyl-CoA into acetyl-CoA through the following reaction:
Reaction by which MDC transforms malonyl-CoA into acetyl-CoA
In the cytosol, malonyl-CoA can inhibit the entrance of fatty acids into the mitochondria and it can also act as a precursor for the fatty acids synthesis. Malonyl-CoA also plays an important role inside the mitochondria, where it is an intermediary between fatty acids and acetyl-CoA, which will be a reserve for the Krebs cycle.
Cytoplasmic MCD is thought to play a role in the regulation of cytoplasmic malonyl-CoA abundance and, therefore, of mitochondrial fatty acid uptake and oxidation.[9] It has been observed that MCD mRNA is most abundant in cardiac and skeletal muscles, tissues in which cytoplasmic malonyl-CoA is a strong inhibitor of mitochondrial fatty acid oxidation and which derive significant amounts of energy from fatty acid oxidation.
In peroxisomes, it is proposed that this enzyme could be involved in degrading intraperoxisomal malonyl-CoA, which is produced by the peroxisomal beta oxidation of odd chain length dicarboxylic fatty acids (odd chain length DFAs). While long and medium chain fatty acids are oxidized mainly in the mitochondria, DFAs are oxidized primarily in peroxisomes, which degrade DFAs completely to malonyl-CoA (in the case of odd chain length DFAs) and oxalyl-CoA (for even chain length DFAs). The peroxisomal form of MCD could function to eliminate this final malonyl-CoA.
Malonyl-CoA acts as an intermediary between fatty acids and acetyl-CoA in the mitochondria, where MCD is believed to participate in the elimination of the residual malonyl-CoA, so that acetyl-CoA can enter the Krebs cycle.
MCD also plays a role in the regulation of glucose and lipids as fuels in human tissues. Malonyl-CoA concentrations are crucial in the intracellular energetic regulation and the production or degradation of this metabolite delimits the use of glucose or lipids to produce ATP.
Pathology
The diseases related with MCD can be caused by its mislocalization, mutations affecting the gene MLYCD, its accumulation in peroxisomes and, mainly, its deficiency.
MCS deficiency is a rare autosomal disorder that is widely diagnosed by neonatal screening and it is caused by mutations in MLYCD. It causes many symptoms: brain abnormalities, mild mental retardation, seizures, hypotonia, metabolic acidosis, vomiting, excretion of malonic and methylmalonic acids in urine, cardiomyopathies, and hypoglycemia. More rarely, it can cause rheumatoid arthritis too.
In peroxisomes, the accumulation of MCD substance also causes pathological symptoms, which are similar to MCS deficiency: malonic aciduria, a lethal disease in which patients (normally children) have delayed development and can suffer from seizures, diarrhoea, hypoglycaemia and cardiomyopathy, as well.
Others symptoms caused by an altered action of MCD can be abdominal pain and chronic constipation.[10]
Localization
Malonyl-CoA decarboxylase is present in the cytosolic, mitochondrial and peroxisomal compartments. MCD is found from bacteria to plants.[11][12] In humans, MCD has been identified in heart, skeletal tissue, pancreas and kidneys. In rats, MCD has been detected in fat, heart and liver.[13]
Enzyme regulation
Because the formation of interchain disulfide bonds leads to positive cooperativity between active sites and increases the affinity for malonyl-CoA and the catalytic efficiency (in vitro), MCD activity doesn't need the intervention of any cofactors or divalent metal ions.[14]
Medical applications
MCD is involved in regulating cardiac malonyl-CoA levels, inhibition of MCD can limit rates of fatty acid oxidation, leading to a secondary increase in glucose oxidation associated with an improvement in the functional recovery of the heart during ischaemia/reperfusion injury. MCD is a potential novel target for cancer treatment.
Related Research Articles
Acetyl-CoA is a molecule that participates in many biochemical reactions in protein, carbohydrate and lipid metabolism. Its main function is to deliver the acetyl group to the citric acid cycle to be oxidized for energy production. Coenzyme A consists of a β-mercaptoethylamine group linked to the vitamin pantothenic acid (B5) through an amide linkage and 3'-phosphorylated ADP. The acetyl group of acetyl-CoA is linked to the sulfhydryl substituent of the β-mercaptoethylamine group. This thioester linkage is a "high energy" bond, which is particularly reactive. Hydrolysis of the thioester bond is exergonic (−31.5 kJ/mol).
Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria. In support of energy metabolism, carnitine transports long-chain fatty acids from the cytosol into mitochondria to be oxidized for free energy production, and also participates in removing products of metabolism from cells. Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source. Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.
Ketogenesis is the biochemical process through which organisms produce ketone bodies by breaking down fatty acids and ketogenic amino acids. The process supplies energy to certain organs, particularly the brain, heart and skeletal muscle, under specific scenarios including fasting, caloric restriction, sleep, or others.
Enoyl-CoA-(∆) isomerase (EC 5.3.3.8, also known as dodecenoyl-CoA- isomerase, 3,2-trans-enoyl-CoA isomerase, ∆3 ,∆2 -enoyl-CoA isomerase, or acetylene-allene isomerase, is an enzyme that catalyzes the conversion of cis- or trans-double bonds of coenzyme A bound fatty acids at gamma-carbon to trans double bonds at beta-carbon as below:
Oxaloacetic acid (also known as oxalacetic acid or OAA) is a crystalline organic compound with the chemical formula HO2CC(O)CH2CO2H. Oxaloacetic acid, in the form of its conjugate base oxaloacetate, is a metabolic intermediate in many processes that occur in animals. It takes part in gluconeogenesis, the urea cycle, the glyoxylate cycle, amino acid synthesis, fatty acid synthesis and the citric acid cycle.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA, which enters the citric acid cycle, and NADH and FADH2, which are co-enzymes used in the electron transport chain. It is named as such because the beta carbon of the fatty acid undergoes oxidation to a carbonyl group. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
Acetyl-CoA carboxylase (ACC) is a biotin-dependent enzyme that catalyzes the irreversible carboxylation of acetyl-CoA to produce malonyl-CoA through its two catalytic activities, biotin carboxylase (BC) and carboxyltransferase (CT). ACC is a multi-subunit enzyme in most prokaryotes and in the chloroplasts of most plants and algae, whereas it is a large, multi-domain enzyme in the cytoplasm of most eukaryotes. The most important function of ACC is to provide the malonyl-CoA substrate for the biosynthesis of fatty acids. The activity of ACC can be controlled at the transcriptional level as well as by small molecule modulators and covalent modification. The human genome contains the genes for two different ACCs—ACACA and ACACB.
Malonyl-CoA is a coenzyme A derivative of malonic acid.
Malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.
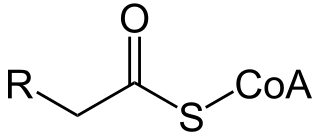
Acyl-CoA is a group of coenzymes that metabolize fatty acids. Acyl-CoA's are susceptible to beta oxidation, forming, ultimately, acetyl-CoA. The acetyl-CoA enters the citric acid cycle, eventually forming several equivalents of ATP. In this way, fats are converted to ATP, the universal biochemical energy carrier.
In biochemistry, fatty acid synthesis is the creation of fatty acids from acetyl-CoA and NADPH through the action of enzymes called fatty acid synthases. This process takes place in the cytoplasm of the cell. Most of the acetyl-CoA which is converted into fatty acids is derived from carbohydrates via the glycolytic pathway. The glycolytic pathway also provides the glycerol with which three fatty acids can combine to form triglycerides, the final product of the lipogenic process. When only two fatty acids combine with glycerol and the third alcohol group is phosphorylated with a group such as phosphatidylcholine, a phospholipid is formed. Phospholipids form the bulk of the lipid bilayers that make up cell membranes and surrounds the organelles within the cells.
Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine, but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.
2,4 Dienoyl-CoA reductase also known as DECR1 is an enzyme which in humans is encoded by the DECR1 gene which resides on chromosome 8. This enzyme catalyzes the following reactions
Thiolases, also known as acetyl-coenzyme A acetyltransferases (ACAT), are enzymes which convert two units of acetyl-CoA to acetoacetyl CoA in the mevalonate pathway.
The Randle cycle, also known as the glucose fatty-acid cycle, is a metabolic process involving the competition of glucose and fatty acids for substrates. It is theorized to play a role in explaining type 2 diabetes and insulin resistance.
Carnitine O-octanoyltransferase is a member of the transferase family, more specifically a carnitine acyltransferase, a type of enzyme which catalyzes the transfer of acyl groups from acyl-CoAs to carnitine, generating CoA and an acyl-carnitine. The systematic name of this enzyme is octanoyl-CoA:L-carnitine O-octanoyltransferase. Other names in common use include medium-chain/long-chain carnitine acyltransferase, carnitine medium-chain acyltransferase, easily solubilized mitochondrial carnitine palmitoyltransferase, and overt mitochondrial carnitine palmitoyltransferase. Specifically, CROT catalyzes the chemical reaction:
Acyl-CoA thioesterase 2, also known as ACOT2, is an enzyme which in humans is encoded by the ACOT2 gene.
Acyl-CoA synthetase family member 3 is an enzyme that in humans is encoded by the ACSF3 gene.
The citrate-malate shuttle is a series of chemical reactions – commonly referred to as a biochemical cycle or system – that transports acetyl-CoA in the mitochondrial matrix across the inner and outer mitochondrial membrane for fatty acid synthesis. Mitochondria is enclosed in a double membrane. As the inner mitochondrial membrane is impermeable to acetyl-CoA, the shuttle system is essential to fatty acid synthesis in the cytosol. It plays an important role in the generation of lipids in the liver.
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid. Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism. Due to being infrequently diagnosed, it most often goes undetected.
↑ Aparicio Alarcón, David (April 5, 2013). Estudio estructural y funcional de malonil-CoA descarboxilasa humana, un enzima peroxisomal clave en la regulación de malonil-CoA y ácidos grasos (Thesis) (in Spanish). Universitat Autònoma de Barcelona. hdl:10803/113486.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.