Sly syndrome, also called mucopolysaccharidosis type VII (MPS-VII), is an autosomal recessive lysosomal storage disease caused by a deficiency of the enzyme β-glucuronidase. This enzyme is responsible for breaking down large sugar molecules called glycosaminoglycans. The inability to break down GAGs leads to a buildup in many tissues and organs of the body. The severity of the disease can vary widely.

Mucopolysaccharidoses are a group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules called glycosaminoglycans (GAGs). These long chains of sugar carbohydrates occur within the cells that help build bone, cartilage, tendons, corneas, skin and connective tissue. GAGs are also found in the fluids that lubricate joints.

Lysosomal storage diseases are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function. Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.

Sanfilippo syndrome, also known as mucopolysaccharidosis type III (MPS III), is a rare autosomal recessive lysosomal storage disease that primarily affects the brain and spinal cord. It is caused by a buildup of large sugar molecules called glycosaminoglycans (GAGs, or mucopolysaccharides) in the body's lysosomes.
Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.

Hurler syndrome, also known as mucopolysaccharidosis Type IH (MPS-IH), Hurler's disease, and formerly gargoylism, is a genetic disorder that results in the buildup of large sugar molecules called glycosaminoglycans (GAGs) in lysosomes. The inability to break down these molecules results in a wide variety of symptoms caused by damage to several different organ systems, including but not limited to the nervous system, skeletal system, eyes, and heart.

Morquio syndrome, also known as mucopolysaccharidosis type IV (MPS IV), is a rare metabolic disorder in which the body cannot process certain types of sugar molecules called glycosaminoglycans (AKA GAGs, or mucopolysaccharides). In Morquio syndrome, the specific GAG which builds up in the body is called keratan sulfate. This birth defect, which is autosomal recessive, is a type of lysosomal storage disorder. The buildup of GAGs in different parts of the body causes symptoms in many different organ systems. In the US, the incidence rate for Morquio syndrome is estimated at between 1 in 200,000 and 1 in 300,000 live births.

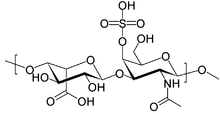
Hunter syndrome, or mucopolysaccharidosis type II, is a rare genetic disorder in which large sugar molecules called glycosaminoglycans build up in body tissues. It is a form of lysosomal storage disease. Hunter syndrome is caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (I2S). The lack of this enzyme causes heparan sulfate and dermatan sulfate to accumulate in all body tissues. Hunter syndrome is the only MPS syndrome to exhibit X-linked recessive inheritance.
The GM1 gangliosidoses, usually shortened to GM1, are gangliosidoses caused by mutation in the GLB1 gene resulting in a deficiency of beta-galactosidase. The deficiency causes abnormal storage of acidic lipid materials in cells of the central and peripheral nervous systems, but particularly in the nerve cells, resulting in progressive neurodegeneration. GM1 is a rare lysosomal storage disorder with a prevalence of 1 to every 100,000 to 200,000 live births worldwide, although rates are higher in some regions.

Pseudo-Hurler polydystrophy, also referred to as mucolipidosis III, is a lysosomal storage disease closely related to I-cell disease. This disorder is called Pseudo-Hurler because it resembles a mild form of Hurler syndrome, one of the mucopolysaccharide (MPS) diseases.
Inclusion-cell (I-cell) disease, also referred to as mucolipidosis II, is part of the lysosomal storage disease family and results from a defective phosphotransferase. This enzyme transfers phosphate to mannose residues on specific proteins. Mannose-6-phosphate serves as a marker for proteins to be targeted to lysosomes within the cell. Without this marker, proteins are instead secreted outside the cell, which is the default pathway for proteins moving through the Golgi apparatus. Lysosomes cannot function without these proteins, which function as catabolic enzymes for the normal breakdown of substances in various tissues throughout the body. As a result, a buildup of these substances occurs within lysosomes because they cannot be degraded, resulting in the characteristic I-cells, or "inclusion cells" seen microscopically. In addition, the defective lysosomal enzymes normally found only within lysosomes are instead found in high concentrations in the blood, but they remain inactive at blood pH because they require the low lysosomal pH 5 to function.
An osteochondrodysplasia, or skeletal dysplasia, is a disorder of the development of bone and cartilage. Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. These disorders lead to disproportionate short stature and bone abnormalities, particularly in the arms, legs, and spine. Skeletal dysplasia can result in marked functional limitation and even mortality.
Iduronidase, sold as Aldurazyme, is an enzyme with the systematic name glycosaminoglycan α-L-iduronohydrolase. It catalyses the hydrolysis of unsulfated α-L-iduronosidic linkages in dermatan sulfate.

Arylsulfatase B is an enzyme associated with mucopolysaccharidosis VI.
Maroteaux–Lamy syndrome, or Mucopolysaccharidosis Type VI (MPS-VI), is an inherited disease caused by a deficiency in the enzyme arylsulfatase B (ARSB). ASRB is responsible for the breakdown of large sugar molecules called glycosaminoglycans. In particular, ARSB breaks down dermatan sulfate and chondroitin sulfate. Because people with MPS-VI lack the ability to break down these GAGs, these chemicals build up in the lysosomes of cells. MPS-VI is therefore a type of lysosomal storage disease.

Scheie syndrome is a disease caused by a deficiency in the enzyme iduronidase, leading to the buildup of glycosaminoglycans (GAGs) in the body. It is the most mild subtype of mucopolysaccharidosis type I; the most severe subtype of this disease is called Hurler Syndrome.

Hurler–Scheie syndrome is a genetic disorder caused by the buildup of glycosaminoglycans (GAGs) in various organ tissues. It is a cutaneous condition, also characterized by mild mental retardation and corneal clouding. Respiratory problems, sleep apnea, and heart disease may develop in adolescence.

N-sulphoglucosamine sulphohydrolase is an enzyme that in humans is encoded by the SGSH gene.

Galactosialidosis, also known as neuraminidase deficiency with beta-galactosidase deficiency, is a genetic lysosomal storage disease. It is caused by a mutation in the CTSA gene which leads to a deficiency of enzymes β-galactosidase and neuraminidase. This deficiency inhibits the lysosomes of cells from functioning properly, resulting in the accumulation of toxic matter within the cell. Hallmark symptoms include abnormal spinal structure, vision problems, coarse facial features, hearing impairment, and intellectual disability. Because galactosialidosis involves the lysosomes of all cells, it can affect various areas of the body, including the brain, eyes, bones, and muscles. Depending on the patient's age at the onset of symptoms, the disease consists of three subtypes: early infantile, late infantile, and juvenile/adult. This condition is considered rare, with most cases having been in the juvenile/adult group of patients.
Coarse facial features is a constellation of facial features that are present in many inborn errors of metabolism.