In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
In chemistry a one-pot synthesis is a strategy to improve the efficiency of a chemical reaction in which a reactant is subjected to successive chemical reactions in just one reactor. This is much desired by chemists because avoiding a lengthy separation process and purification of the intermediate chemical compounds can save time and resources while increasing chemical yield.
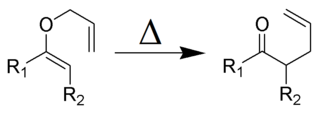
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation.

The Curtius rearrangement, first defined by Theodor Curtius in 1885, is the thermal decomposition of an acyl azide to an isocyanate with loss of nitrogen gas. The isocyanate then undergoes attack by a variety of nucleophiles such as water, alcohols and amines, to yield a primary amine, carbamate or urea derivative respectively. Several reviews have been published.
The Dakin–West reaction is a chemical reaction that transforms an amino-acid into a keto-amide using an acid anhydride and a base, typically pyridine. It is named for Henry Drysdale Dakin (1880–1952) and Randolph West (1890–1949). In 2016 Schreiner and coworkers reported the first asymmetric variant of this reaction employing short oligopeptides as catalysts.

1,1'-Carbonyldiimidazole (CDI) is an organic compound with the molecular formula (C3H3N2)2CO. It is a white crystalline solid. It is often used for the coupling of amino acids for peptide synthesis and as a reagent in organic synthesis.
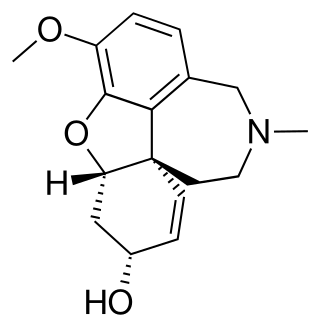
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.

In organic chemistry, aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine and two methylene bridges. The parent compound is aziridine, with molecular formula C2H4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
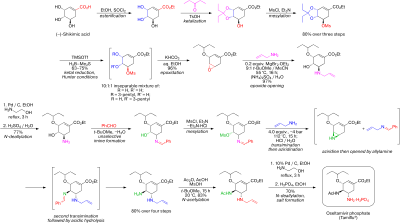
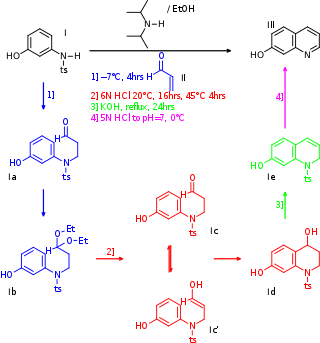
The total synthesis of quinine, a naturally-occurring antimalarial drug, was developed over a 150-year period. The development of synthetic quinine is considered a milestone in organic chemistry although it has never been produced industrially as a substitute for natural occurring quinine. The subject has also been attended with some controversy: Gilbert Stork published the first stereoselective total synthesis of quinine in 2001, meanwhile shedding doubt on the earlier claim by Robert Burns Woodward and William Doering in 1944, claiming that the final steps required to convert their last synthetic intermediate, quinotoxine, into quinine would not have worked had Woodward and Doering attempted to perform the experiment. A 2001 editorial published in Chemical & Engineering News sided with Stork, but the controversy was eventually laid to rest once and for all when Williams and coworkers successfully repeated Woodward's proposed conversion of quinotoxine to quinine in 2007.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being awarded one half of the 2001 Nobel Prize in Chemistry.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.

Allyl glycidyl ether is an organic compound used in adhesives and sealants and as a monomer for polymerization reactions. It is formally the condensation product of allyl alcohol and glycidol via an ether linkage. Because it contains both an alkene and an epoxide group, either group can be reacted selectively to yield a product where the other functional group remains intact for future reactions.

The Krische allylation involves the enantioselective iridium-catalyzed addition of an allyl group to an aldehyde or an alcohol, resulting in the formation of a secondary homoallylic alcohol. The mechanism of the Krische allylation involves primary alcohol dehydrogenation or, when using aldehyde reactants, hydrogen transfer from 2-propanol. Unlike other allylation methods, the Krische allylation avoids the use of preformed allyl metal reagents and enables the direct conversion of primary alcohols to secondary homoallylic alcohols.
In organic chemistry, the Roskamp reaction is a name reaction describing the reaction between α-diazoesters (such as ethyl diazoacetate) and aldehydes to form β-ketoesters, often utilizing various Lewis acids (such as BF3, SnCl2, and GeCl2) as catalysts. The reaction is notable for its mild reaction conditions and selectivity.

Iodine azide is an explosive inorganic compound, which in ordinary conditions is a yellow solid. Formally, it is an inter-pseudohalogen.

The nitro-Mannich reaction is the nucleophilic addition of a nitroalkane to an imine, resulting in the formation of a beta-nitroamine. With the reaction involving the addition of an acidic carbon nucleophile to a carbon-heteroatom double bond, the nitro-Mannich reaction is related to some of the most fundamental carbon-carbon bond forming reactions in organic chemistry, including the aldol reaction, Henry reaction and Mannich reaction.
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.