Hereditary coproporphyria (HCP) is a disorder of heme biosynthesis, classified as an acute hepatic porphyria. HCP is caused by a deficiency of the enzyme coproporphyrinogen oxidase, coded for by the CPOX gene, and is inherited in an autosomal dominant fashion, although homozygous individuals have been identified. Unlike acute intermittent porphyria, individuals with HCP can present with cutaneous findings similar to those found in porphyria cutanea tarda in addition to the acute attacks of abdominal pain, vomiting and neurological dysfunction characteristic of acute porphyrias. Like other porphyrias, attacks of HCP can be induced by certain drugs, environmental stressors or diet changes. Biochemical and molecular testing can be used to narrow down the diagnosis of a porphyria and identify the specific genetic defect. Overall, porphyrias are rare diseases. The combined incidence for all forms of the disease has been estimated at 1:20,000. The exact incidence of HCP is difficult to determine, due to its reduced penetrance.

Variegate porphyria, also known by several other names, is an autosomal dominant porphyria that can have acute symptoms along with symptoms that affect the skin. The disorder results from low levels of the enzyme responsible for the seventh step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.
Acute intermittent porphyria (AIP) is a rare metabolic disorder affecting the production of heme resulting from a deficiency of the enzyme porphobilinogen deaminase. It is the most common of the acute porphyrias.

Protoporphyrinogen oxidase or protox is an enzyme that in humans is encoded by the PPOX gene.

Uroporphyrinogen III decarboxylase is an enzyme that in humans is encoded by the UROD gene.
Erythropoietic porphyria is a type of porphyria associated with erythropoietic cells. In erythropoietic porphyrias, the enzyme deficiency occurs in the red blood cells.

Cystathionine-β-synthase, also known as CBS, is an enzyme (EC 4.2.1.22) that in humans is encoded by the CBS gene. It catalyzes the first step of the transsulfuration pathway, from homocysteine to cystathionine:

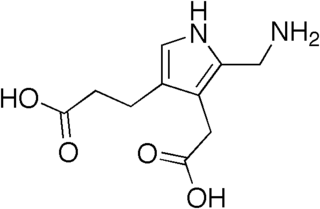
Aminolevulinic acid dehydratase (porphobilinogen synthase, or ALA dehydratase, or aminolevulinate dehydratase) is an enzyme (EC 4.2.1.24) that in humans is encoded by the ALAD gene. Porphobilinogen synthase (or ALA dehydratase, or aminolevulinate dehydratase) synthesizes porphobilinogen through the asymmetric condensation of two molecules of aminolevulinic acid. All natural tetrapyrroles, including hemes, chlorophylls and vitamin B12, share porphobilinogen as a common precursor. Porphobilinogen synthase is the prototype morpheein.

Usherin is a protein that in humans is encoded by the USH2A gene.

Glucose-6-phosphatase, catalytic subunit is an enzyme that in humans is encoded by the G6PC gene.

Exostosin-1 is a protein that in humans is encoded by the EXT1 gene.

6-pyruvoyltetrahydropterin synthase, also known as PTS, is a human gene which facilitates folate biosynthesis.

Beta-sarcoglycan is a protein that in humans is encoded by the SGCB gene.

Phosphorylase b kinase regulatory subunit alpha, liver isoform is an enzyme that in humans is encoded by the PHKA2 gene.

Molybdenum cofactor biosynthesis protein 1 is a protein that in humans and other animals, fungi, and cellular slime molds, is encoded by the MOCS1 gene.

Molybdenum cofactor synthesis protein 2A and molybdenum cofactor synthesis protein 2B are a pair of proteins that in humans are encoded from the same MOCS2 gene. These two proteins dimerize to form molybdopterin synthase.

Y+L amino acid transporter 1 is a protein that in humans is encoded by the SLC7A7 gene.

Adenylyltransferase and sulfurtransferase MOCS3 is an enzyme that in humans is encoded by the MOCS3 gene.

UDP-glucuronosyltransferase 1-9 is an enzyme that in humans is encoded by the UGT1A9 gene.

Urs Albert Meyer is a Swiss physician-scientist and clinical pharmacologist.