A semantic network, or frame network is a knowledge base that represents semantic relations between concepts in a network. This is often used as a form of knowledge representation. It is a directed or undirected graph consisting of vertices, which represent concepts, and edges, which represent semantic relations between concepts, mapping or connecting semantic fields.
SOAP is a messaging protocol specification for exchanging structured information in the implementation of web services in computer networks. Its purpose is to provide extensibility, neutrality and independence. It uses XML Information Set for its message format, and relies on application layer protocols, most often Hypertext Transfer Protocol (HTTP) or Simple Mail Transfer Protocol (SMTP), for message negotiation and transmission.

In computer science, a tree is a widely used abstract data type (ADT)—or data structure implementing this ADT—that simulates a hierarchical tree structure, with a root value and subtrees of children with a parent node, represented as a set of linked nodes.

A phylogenetic tree or evolutionary tree is a branching diagram or "tree" showing the evolutionary relationships among various biological species or other entities—their phylogeny —based upon similarities and differences in their physical or genetic characteristics. All life on Earth is part of a single phylogenetic tree, indicating common ancestry.

A distributed hash table (DHT) is a class of a decentralized distributed system that provides a lookup service similar to a hash table: pairs are stored in a DHT, and any participating node can efficiently retrieve the value associated with a given key. Keys are unique identifiers which map to particular values, which in turn can be anything from addresses, to documents, to arbitrary data. Responsibility for maintaining the mapping from keys to values is distributed among the nodes, in such a way that a change in the set of participants causes a minimal amount of disruption. This allows a DHT to scale to extremely large numbers of nodes and to handle continual node arrivals, departures, and failures.
In mathematics, a binary relation R over a set X is reflexive if every element of X is related to itself. Formally, this may be written ∀x ∈ X : x R x.
In mathematics, the transitive closure of a binary relation R on a set X is the smallest relation on X that contains R and is transitive.
Social network analysis (SNA) is the process of investigating social structures through the use of networks and graph theory. It characterizes networked structures in terms of nodes and the ties, edges, or links that connect them. Examples of social structures commonly visualized through social network analysis include social media networks, memes spread, information circulation, friendship and acquaintance networks, business networks, social networks, collaboration graphs, kinship, disease transmission, and sexual relationships. These networks are often visualized through sociograms in which nodes are represented as points and ties are represented as lines. These visualizations provide a means of qualitatively assessing networks by varying the visual representation of their nodes and edges to reflect attributes of interest.
This is a glossary of graph theory terms. Graph theory is the study of graphs, systems of nodes or vertices connected in pairs by edges.
In mathematics, specifically order theory, a well-quasi-ordering or wqo is a quasi-ordering such that any infinite sequence of elements , , , … from contains an increasing pair with .
In computer science, a topological sort or topological ordering of a directed graph is a linear ordering of its vertices such that for every directed edge uv from vertex u to vertex v, u comes before v in the ordering. For instance, the vertices of the graph may represent tasks to be performed, and the edges may represent constraints that one task must be performed before another; in this application, a topological ordering is just a valid sequence for the tasks. A topological ordering is possible if and only if the graph has no directed cycles, that is, if it is a directed acyclic graph (DAG). Any DAG has at least one topological ordering, and algorithms are known for constructing a topological ordering of any DAG in linear time.
Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. For example, these techniques have been used to explore the family tree of hominid species and the relationships between specific genes shared by many types of organisms. Traditional phylogenetics relies on morphological data obtained by measuring and quantifying the phenotypic properties of representative organisms, while the more recent field of molecular phylogenetics uses nucleotide sequences encoding genes or amino acid sequences encoding proteins as the basis for classification. Many forms of molecular phylogenetics are closely related to and make extensive use of sequence alignment in constructing and refining phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The phylogenetic trees constructed by computational methods are unlikely to perfectly reproduce the evolutionary tree that represents the historical relationships between the species being analyzed. The historical species tree may also differ from the historical tree of an individual homologous gene shared by those species.
Quasi-set theory is a formal mathematical theory for dealing with collections of indistinguishable objects, mainly motivated by the assumption that certain objects treated in quantum physics are indistinguishable and don't have individuality.
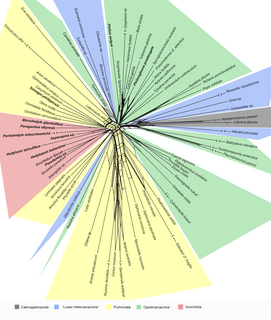
SplitsTree is a popular program for inferring phylogenetic trees or, more generally, phylogenetic networks from various types of data such as a sequence alignment, a distance matrix or a set of trees. SplitsTree implements published methods such as split decomposition neighbor-net, consensus networks, super networks methods or methods for computing hybridization or simple recombination networks.
Triadic closure is a concept in social network theory, first suggested by German sociologist Georg Simmel in his 1908 book Soziologie [Sociology: Investigations on the Forms of Sociation]. Triadic closure is the property among three nodes A, B, and C, such that if a strong tie exists between A-B and A-C, there is a weak or strong tie between B-C. This property is too extreme to hold true across very large, complex networks, but it is a useful simplification of reality that can be used to understand and predict networks.
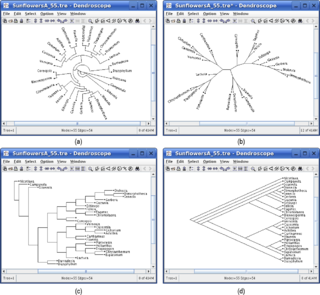
Dendroscope is an interactive computer software program written in Java for viewing Phylogenetic trees. This program is designed to view trees of all sizes and is very useful for creating figures. Dendroscope can be used for a variety of analyses of molecular data sets but is particularly designed for metagenomics or analyses of uncultured environmental samples.

In graph theory, a division of mathematics, a median graph is an undirected graph in which every three vertices a, b, and c have a unique median: a vertex m(a,b,c) that belongs to shortest paths between each pair of a, b, and c.

