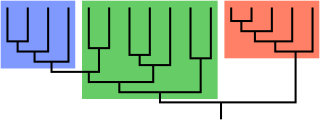
In biological phylogenetics, a clade, also known as a monophyletic group or natural group, is a grouping of organisms that are monophyletic – that is, composed of a common ancestor and all its lineal descendants – on a phylogenetic tree. In the taxonomical literature, sometimes the Latin form cladus is used rather than the English form. Clades are the fundamental unit of cladistics, a modern approach to taxonomy adopted by most biological fields.
In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference, methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.
A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time. In other words, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. In evolutionary biology, all life on Earth is theoretically part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the study of phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.
In phylogenetics and computational phylogenetics, maximum parsimony is an optimality criterion under which the phylogenetic tree that minimizes the total number of character-state changes. Under the maximum-parsimony criterion, the optimal tree will minimize the amount of homoplasy. In other words, under this criterion, the shortest possible tree that explains the data is considered best. Some of the basic ideas behind maximum parsimony were presented by James S. Farris in 1970 and Walter M. Fitch in 1971.
In phylogenetics, long branch attraction (LBA) is a form of systematic error whereby distantly related lineages are incorrectly inferred to be closely related. LBA arises when the amount of molecular or morphological change accumulated within a lineage is sufficient to cause that lineage to appear similar to another long-branched lineage, solely because they have both undergone a large amount of change, rather than because they are related by descent. Such bias is more common when the overall divergence of some taxa results in long branches within a phylogeny. Long branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched. The frequency of true LBA is unclear and often debated, and some authors view it as untestable and therefore irrelevant to empirical phylogenetic inference. Although often viewed as a failing of parsimony-based methodology, LBA could in principle result from a variety of scenarios and be inferred under multiple analytical paradigms.
A phylogenetic network is any graph used to visualize evolutionary relationships between nucleotide sequences, genes, chromosomes, genomes, or species. They are employed when reticulation events such as hybridization, horizontal gene transfer, recombination, or gene duplication and loss are believed to be involved. They differ from phylogenetic trees by the explicit modeling of richly linked networks, by means of the addition of hybrid nodes instead of only tree nodes. Phylogenetic trees are a subset of phylogenetic networks. Phylogenetic networks can be inferred and visualised with software such as SplitsTree, the R-package, phangorn, and, more recently, Dendroscope. A standard format for representing phylogenetic networks is a variant of Newick format which is extended to support networks as well as trees.
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.
The extensible NEXUS file format is widely used in bioinformatics. It stores information about taxa, morphological and molecular characters, distances, genetic codes, assumptions, sets, trees, etc. Several popular phylogenetic programs such as PAUP*, MrBayes, Mesquite, MacClade and SplitsTree use this format.
Tree rearrangements are deterministic algorithms devoted to search for optimal phylogenetic tree structure. They can be applied to any set of data that are naturally arranged into a tree, but have most applications in computational phylogenetics, especially in maximum parsimony and maximum likelihood searches of phylogenetic trees, which seek to identify one among many possible trees that best explains the evolutionary history of a particular gene or species.
In mathematics and phylogenetics, Newick tree format is a way of representing graph-theoretical trees with edge lengths using parentheses and commas. It was adopted by James Archie, William H. E. Day, Joseph Felsenstein, Wayne Maddison, Christopher Meacham, F. James Rohlf, and David Swofford, at two meetings in 1986, the second of which was at Newick's restaurant in Dover, New Hampshire, US. The adopted format is a generalization of the format developed by Meacham in 1984 for the first tree-drawing programs in Felsenstein's PHYLIP package.

SplitsTree is a popular freeware program for inferring phylogenetic trees, phylogenetic networks, or, more generally, splits graphs, from various types of data such as a sequence alignment, a distance matrix or a set of trees. SplitsTree implements published methods such as split decomposition, neighbor-net, consensus networks, super networks methods or methods for computing hybridization or simple recombination networks. It uses the NEXUS file format. The splits graph is defined using a special data block.
A split in phylogenetics is a bipartition of a set of taxa, and the smallest unit of information in unrooted phylogenetic trees: each edge of an unrooted phylogenetic tree represents one split, and the tree can be efficiently reconstructed from its set of splits. Moreover, when given several trees, the splits occurring in more than half of these trees give rise to a consensus tree, and the splits occurring in a smaller fraction of the trees generally give rise to a consensus split network.
Distance matrices are used in phylogeny as non-parametric distance methods and were originally applied to phenetic data using a matrix of pairwise distances. These distances are then reconciled to produce a tree. The distance matrix can come from a number of different sources, including measured distance or morphometric analysis, various pairwise distance formulae applied to discrete morphological characters, or genetic distance from sequence, restriction fragment, or allozyme data. For phylogenetic character data, raw distance values can be calculated by simply counting the number of pairwise differences in character states.
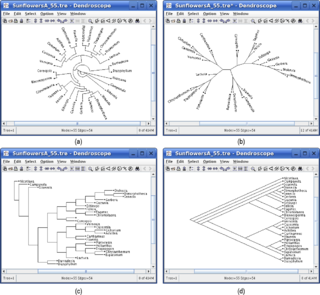
Dendroscope is an interactive computer software program written in Java for viewing Phylogenetic trees. This program is designed to view trees of all sizes and is very useful for creating figures. Dendroscope can be used for a variety of analyses of molecular data sets but is particularly designed for metagenomics or analyses of uncultured environmental samples.
Quantitative comparative linguistics is the use of quantitative analysis as applied to comparative linguistics. Examples include the statistical fields of lexicostatistics and glottochronology, and the borrowing of phylogenetics from biology.
The Robinson–Foulds or symmetric difference metric, often abbreviated as the RF distance, is a simple way to calculate the distance between phylogenetic trees.

In mathematics and computer science, an unrooted binary tree is an unrooted tree in which each vertex has either one or three neighbors.
The concept of a quasi-median network is a generalization of the concept of a median network that was introduced to represent multistate characters. Note that, unlike median networks, quasi-median networks are not split networks. A quasi-median network is defined as a phylogenetic network, the node set of which is given by the quasi-median closure of the condensed version of M and in which any two nodes are joined by an edge if and only if the sequences associated with the nodes differ in exactly one position. The quasi-median closure is defined as the set of all sequences that can be obtained by repeatedly taking the quasi-median of any three sequences in the set and then adding the result to the set.
In the mathematical field of graph theory, an agreement forest for two given trees is any forest which can, informally speaking, be obtained from both trees by removing a common number of edges.

In phylogenetics, reconciliation is an approach to connect the history of two or more coevolving biological entities. The general idea of reconciliation is that a phylogenetic tree representing the evolution of an entity can be drawn within another phylogenetic tree representing an encompassing entity to reveal their interdependence and the evolutionary events that have marked their shared history. The development of reconciliation approaches started in the 1980s, mainly to depict the coevolution of a gene and a genome, and of a host and a symbiont, which can be mutualist, commensalist or parasitic. It has also been used for example to detect horizontal gene transfer, or understand the dynamics of genome evolution.