A carboxylic acid is an organic acid that contains a carboxyl group (C(=O)OH) attached to an R-group. The general formula of a carboxylic acid is R−COOH or R−CO2H, with R referring to the alkyl, alkenyl, aryl, or other group. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
An ester is a chemical compound derived from an acid in which at least one –OH hydroxyl group is replaced by an –O– alkyl (alkoxy) group, as in the substitution reaction of a carboxylic acid and an alcohol. Glycerides are fatty acid esters of glycerol; they are important in biology, being one of the main classes of lipids and comprising the bulk of animal fats and vegetable oils.
In chemistry, a ketone is a functional group with the structure R2C=O, where R can be a variety of carbon-containing substituents. Ketones contain a carbonyl group (a carbon-oxygen double bond). The simplest ketone is acetone (R = R' = methyl), with the formula CH3C(O)CH3. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids (e.g., testosterone), and the solvent acetone.
Chemically, an aldehyde is a compound containing a functional group with the structure −CHO, consisting of a carbonyl center with the carbon atom also bonded to hydrogen and to any generic alkyl or side chain R group. The functional group itself is known as an aldehyde or formyl group.

An acetal is a functional group with the connectivity R2C(OR')2). Here, the R groups can be organic fragments (a carbon atom, with arbitrary other atoms attached to that) or hydrogen, while the R' groups must be organic fragments not hydrogen. The two R' groups can be equivalent to each other (a "symmetric acetal") or not (a "mixed acetal"). Acetals are formed from and convertible to aldehydes or ketones and have the same oxidation state at the central carbon, but have substantially different chemical stability and reactivity as compared to the analogous carbonyl compounds. The central carbon atom has four bonds to it, and is therefore saturated and has tetrahedral geometry.

In organic chemistry, a carbonyl group is a functional group composed of a carbon atom double-bonded to an oxygen atom: C=O. It is common to several classes of organic compounds, as part of many larger functional groups. A compound containing a carbonyl group is often referred to as a carbonyl compound.

The aldol reaction is a means of forming carbon–carbon bonds in organic chemistry. Discovered independently by the Russian chemist Alexander Borodin in 1869 and by the French chemist Charles-Adolphe Wurtz in 1872, the reaction combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. These products are known as aldols, from the aldehyde + alcohol, a structural motif seen in many of the products. Aldol structural units are found in many important molecules, whether naturally occurring or synthetic. For example, the aldol reaction has been used in the large-scale production of the commodity chemical pentaerythritol and the synthesis of the heart disease drug Lipitor.
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electrophilic double or triple bond reacts with a nucleophile, such that the double or triple bond is broken. Nucleophilic additions differ from electrophilic additions in that the former reactions involve the group to which atoms are added accepting electron pairs, whereas the latter reactions involve the group donating electron pairs.

Organolithium reagents are organometallic compounds that contain carbon – lithium bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C-Li bond is highly ionic. Owing to the polar nature of the C-Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
In organic chemistry, an acyl chloride (or acid chloride) is an organic compound with the functional group -COCl. Their formula is usually written RCOCl, where R is a side chain. They are reactive derivatives of carboxylic acids. A specific example of an acyl chloride is acetyl chloride, CH3COCl. Acyl chlorides are the most important subset of acyl halides.
Ozonolysis is an organic reaction where the unsaturated bonds of alkenes, alkynes, or azo compounds are cleaved with ozone. Alkenes and alkynes form organic compounds in which the multiple carbon–carbon bond has been replaced by a carbonyl group while azo compounds form nitrosamines. The outcome of the reaction depends on the type of multiple bond being oxidized and the work-up conditions.
The Cannizzaro reaction, named after its discoverer Stanislao Cannizzaro, is a chemical reaction which involves the base-induced disproportionation of two molecules of a non-enolizable aldehyde to give a primary alcohol and a carboxylic acid.

Organoborane or organoboron compounds are chemical compounds of boron and carbon that are organic derivatives of BH3, for example trialkyl boranes. Organoboron chemistry or organoborane chemistry is the chemistry of these compounds.
Nucleophilic acyl substitution describe a class of substitution reactions involving nucleophiles and acyl compounds. In this type of reaction, a nucleophile – such as an alcohol, amine, or enolate – displaces the leaving group of an acyl derivative – such as an acid halide, anhydride, or ester. The resulting product is a carbonyl-containing compound in which the nucleophile has taken the place of the leaving group present in the original acyl derivative. Because acyl derivatives react with a wide variety of nucleophiles, and because the product can depend on the particular type of acyl derivative and nucleophile involved, nucleophilic acyl substitution reactions can be used to synthesize a variety of different products.

The Dakin oxidation is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde or ketone reacts with hydrogen peroxide in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidized, and the hydrogen peroxide is reduced.
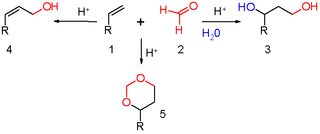
The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol. When water is absent, the cationic intermediate loses a proton to give an allylic alcohol. With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane. When water is replaced by acetic acid the corresponding esters are formed.
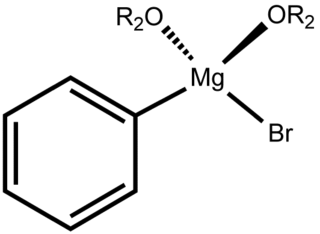
Phenylmagnesium bromide, with the simplified formula C
6H
5MgBr, is a magnesium-containing organometallic compound. It is commercially available as a solution in diethyl ether or tetrahydrofuran (THF). Phenylmagnesium bromide is a Grignard reagent. It is often used as a synthetic equivalent for the phenyl "Ph−" synthon.

Cyclopropanone is an organic compound with molecular formula (CH2)2CO consisting of a cyclopropane carbon framework with a ketone functional group. The parent compound is labile, being highly sensitive toward even weak nucleophiles. Surrogates of cyclopropanone include the ketals.

In organic chemistry, carbonyl reduction is the organic reduction of any carbonyl group by a reducing agent.

The Jones oxidation is an organic reaction for the oxidation of primary and secondary alcohols to carboxylic acids and ketones, respectively. It is named after its discoverer, Sir Ewart Jones. The reaction was an early method for the oxidation of alcohols. Its use has subsided because milder, more selective reagents have been developed, e.g. Collins reagent.