
Gaucher's disease or Gaucher disease (GD) is a genetic disorder in which glucocerebroside accumulates in cells and certain organs. The disorder is characterized by bruising, fatigue, anemia, low blood platelet count and enlargement of the liver and spleen, and is caused by a hereditary deficiency of the enzyme glucocerebrosidase, which acts on glucocerebroside. When the enzyme is defective, glucocerebroside accumulates, particularly in white blood cells and especially in macrophages. Glucocerebroside can collect in the spleen, liver, kidneys, lungs, brain, and bone marrow.

Fabry disease, also known as Anderson–Fabry disease, is a rare genetic disease that can affect many parts of the body, including the kidneys, heart, brain, and skin. Fabry disease is one of a group of conditions known as lysosomal storage diseases. The genetic mutation that causes Fabry disease interferes with the function of an enzyme that processes biomolecules known as sphingolipids, leading to these substances building up in the walls of blood vessels and other organs. It is inherited in an X-linked manner.

Glycogen storage disease type II(GSD-II), also called Pompe disease, and formerly known as GSD-IIa or Limb–girdle muscular dystrophy2V, is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme (GAA). The inability to breakdown glycogen within the lysosomes of cells leads to progressive muscle weakness throughout the body and affects various body tissues, particularly in the heart, skeletal muscles, liver and the nervous system.
Imiglucerase is a medication used in the treatment of Gaucher's disease.

Miglustat, sold under the brand name Zavesca among others, is a medication used to treat type I Gaucher disease and Pompe disease.
Alglucosidase alfa, sold under the brand name Myozyme among others, is an enzyme replacement therapy (ERT) orphan drug for treatment of Pompe disease, a rare lysosomal storage disorder (LSD). Chemically, the drug is an analog of the enzyme that is deficient in patients affected by Pompe disease, alpha-glucosidase. It is the first drug available to treat this disease.
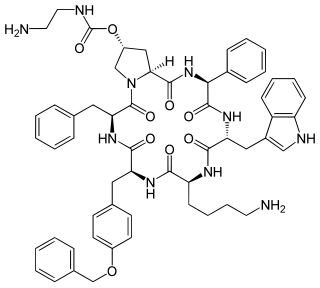
Pasireotide, sold under the brand name Signifor, is an orphan drug approved in the United States and the European Union for the treatment of Cushing's disease in patients who fail or are ineligible for surgical therapy. It was developed by Novartis. Pasireotide is a somatostatin analog with a 40-fold increased affinity to somatostatin receptor 5 compared to other somatostatin analogs.
Sebelipase alfa, sold under the brand name Kanuma, is a recombinant form of the enzyme lysosomal acid lipase (LAL) that is used as a medication for the treatment of lysosomal acid lipase deficiency (LAL-D). It is administered via intraveneous infusion. It was approved for medical use in the European Union and in the United States in 2015.
Asfotase alfa, sold under the brand name Strensiq, is a medication used in the treatment of people with perinatal/infantile- and juvenile-onset hypophosphatasia.

Migalastat, sold under the brand name Galafold, is a medication for the treatment of Fabry disease, a rare genetic disorder. It was developed by Amicus Therapeutics. The US Food and Drug Administration (FDA) granted it orphan drug status in 2004, and the European Commission followed in 2006. The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) granted the drug a marketing approval under the name Galafold in May 2016.
Vestronidase alfa, sold under brand name Mepsevii, is a medication for the treatment of Sly syndrome. It is a recombinant form of the human enzyme beta-glucuronidase. It was approved in the United States in November 2017, to treat children and adults with an inherited metabolic condition called mucopolysaccharidosis type VII, also known as Sly syndrome. MPS VII is an extremely rare, progressive condition that affects most tissues and organs.
Sutimlimab, sold under the brand name Enjaymo, is a monoclonal antibody that is used to treat adults with cold agglutinin disease (CAD). It is given by intravenous infusion. Sutimlimab prevents complement-enhanced activation of autoimmune human B cells in vitro.
Velmanase alfa, sold under the brand name Lamzede, is a medication used for the treatment of alpha-mannosidosis. Velmanase alfa is a recombinant human lysosomal alpha-mannosidase.
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera. It is an interferon. It is given by injection.
Lumasiran, sold under the brand name Oxlumo, is a medication for the treatment of primary hyperoxaluria type 1 (PH1).
Avalglucosidase alfa, sold under the brand name Nexviazyme, is an enzyme replacement therapy medication used for the treatment of glycogen storage disease type II.
Frunevetmab, sold under the brand name Solensia, is a monoclonal antibody used to treat pain associated with osteoarthritis in cats. It is the first monoclonal antibody drug approved by the US Food and Drug Administration for animal use. Frunevetmab is the international nonproprietary name.
Olipudase alfa, sold under the brand name Xenpozyme, is a medication used for the treatment of non-central nervous system (CNS) manifestations of acid sphingomyelinase deficiency type A/B or type B.
Cipaglucosidase alfa, sold under the brand name Pombiliti, and used in combination with miglustat, is a medication used for the treatment of glycogen storage disease type II. Cipaglucosidase alfa is a recombinant human acid α-glucosidase enzyme replacement therapy that provides an exogenous source of acid α-glucosidase.
Pegunigalsidase alfa, sold under the brand name Elfabrio, is an enzyme replacement therapy for the treatment of Fabry disease. It is a recombinant human α-galactosidase-A. It is a hydrolytic lysosomal neutral glycosphingolipid-specific enzyme.