
Blaschko's lines, also called the lines of Blaschko, are lines of normal cell development in the skin. These lines are only visible in those with a mosaic skin condition or in chimeras where different cell lines contain different genes. These lines may express different amounts of melanin, or become visible due to a differing susceptibility to disease. In such individuals, they can become apparent as whorls, patches, streaks or lines in a linear or segmental distribution over the skin. They follow a V shape over the back, S-shaped whirls over the chest and sides, and wavy shapes on the head. Not all mosaic skin conditions follow Blaschko's lines.

Bare lymphocyte syndrome is a condition caused by mutations in certain genes of the major histocompatibility complex or involved with the processing and presentation of MHC molecules. It is a form of severe combined immunodeficiency.
Gray platelet syndrome (GPS), or platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha-granules in blood platelets, and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. The name derives from the initial observation of gray appearance of platelets with a paucity of granules on blood films from a patient with a lifelong bleeding disorder.

RAPADILINO syndrome is an autosomal recessive disorder characterized by:
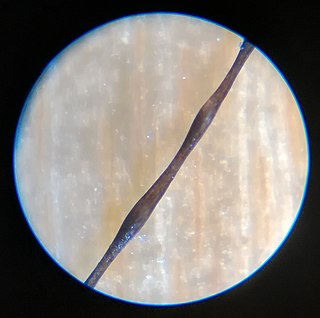
Monilethrix is a rare autosomal dominant hair disease that results in short, fragile, broken hair that appears beaded. It comes from the Latin word for necklace (monile) and the Greek word for hair (thrix). Hair becomes brittle, and breaks off at the thinner parts between the beads. It appears as a thinning or baldness of hair and was first described in 1897 by Walter Smith

Uncombable hair syndrome (UHS) is a rare structural anomaly of the hair with a variable degree of effect. It is characterized by hair that is silvery, dry, frizzy, wiry, and impossible to comb. It was first reported in the early 20th century. It typically becomes apparent between the ages of 3 months and 12 years. UHS has several names, including pili trianguli et canaliculi (Latin), cheveux incoiffables (French), and "spun-glass hair". This disorder is believed to be autosomal recessive in most instances, but there are a few documented cases where multiple family members display the trait in an autosomal dominant fashion. Based on the current scientific studies related to the disorder, the three genes that have been causally linked to UHS are PADI3, TGM3, and TCHH. These genes encode proteins important for hair shaft formation. Clinical symptoms of the disorder arise between 3 months and 12 years of age. The quantity of hair on the head does not change, but hair starts to grow more slowly and becomes increasingly "uncombable". To be clinically apparent, 50% of all scalp hair shafts must be affected by UHS. This syndrome only affects the hair shaft of the scalp and does not influence hair growth in terms of quantity, textural feel, or appearance on the rest of the body.

Zinc transporter ZIP4 is a transmembrane protein which in humans is encoded by the SLC39A4 gene. It is associated with acrodermatitis enteropathica.
Zinc deficiency is defined either as insufficient zinc to meet the needs of the body, or as a serum zinc level below the normal range. However, since a decrease in the serum concentration is only detectable after long-term or severe depletion, serum zinc is not a reliable biomarker for zinc status. Common symptoms include increased rates of diarrhea. Zinc deficiency affects the skin and gastrointestinal tract; brain and central nervous system, immune, skeletal, and reproductive systems.
Gianotti–Crosti syndrome, also known as infantile papular acrodermatitis, papular acrodermatitis of childhood, and papulovesicular acrolocated syndrome, is a reaction of the skin to a viral infection. Hepatitis B virus and Epstein–Barr virus are the most frequently reported pathogens. Other viruses implicated are hepatitis A virus, hepatitis C virus, cytomegalovirus, coxsackievirus, adenovirus, enterovirus, rotavirus, rubella virus, HIV, and parainfluenza virus.
Björnstad syndrome is an autosomal recessive congenital condition involving pili torti, sensorineural deafness, and hair abnormalities. It was first characterized in 1965, in Oslo, by prof. Roar Theodor Bjørnstad after he observed an association between pili torti and hearing loss. The condition is extremely rare, with less than 50 cases documented in medical literature worldwide.
Madarosis is a condition that results in the loss of eyelashes, and sometimes eyebrows. The term "madarosis" is derived from the ancient Greek "madaros", meaning "bald". It originally was a disease of only losing eyelashes but it currently is the loss of both eyelashes and eyebrows. Eyebrows and eyelashes are both important in the prevention of bacteria and other foreign objects from entering the eye. A majority of patients with madarosis have leprosy, and it was reported that 76% of patients with varying types of leprosy had madarosis.
Congenital ichthyosiform erythroderma, also known as nonbullous congenital ichthyosiform erythroderma, is a rare type of the ichthyosis family of skin diseases which occurs in 1 in 200,000 to 300,000 births. The disease comes under the umbrella term autosomal recessive congenital ichthyosis, which include non-syndromic congenital ichthyoses such as harlequin ichthyosis and lamellar ichthyosis.

Aplasia cutis congenita is a rare disorder characterized by congenital absence of skin. Ilona J. Frieden classified ACC in 1986 into 9 groups on the basis of location of the lesions and associated congenital anomalies. The scalp is the most commonly involved area with lesser involvement of trunk and extremities. Frieden classified ACC with fetus papyraceus as type 5. This type presents as truncal ACC with symmetrical absence of skin in stellate or butterfly pattern with or without involvement of proximal limbs. It is the most common congenital cicatricial alopecia, and is a congenital focal absence of epidermis with or without evidence of other layers of the skin.
Trichothiodystrophy (TTD) is an autosomal recessive inherited disorder characterised by brittle hair and intellectual impairment. The word breaks down into tricho – "hair", thio – "sulphur", and dystrophy – "wasting away" or literally "bad nourishment". TTD is associated with a range of symptoms connected with organs of the ectoderm and neuroectoderm. TTD may be subclassified into four syndromes: Approximately half of all patients with trichothiodystrophy have photosensitivity, which divides the classification into syndromes with or without photosensitivity; BIDS and PBIDS, and IBIDS and PIBIDS. Modern covering usage is TTD-P (photosensitive), and TTD.
Junctional epidermolysis bullosa is a skin condition characterized by blister formation within the lamina lucida of the basement membrane zone.
Wrinkly skin syndrome(WSS) is a rare genetic condition characterized by sagging, wrinkled skin, low skin elasticity, and delayed fontanelle (soft spot) closure, along with a range of other symptoms. The disorder exhibits an autosomal recessive inheritance pattern with mutations in the ATP6V0A2 gene, leading to abnormal glycosylation events. There are only about 30 known cases of WSS as of 2010. Given its rarity and symptom overlap with other dermatological conditions, reaching an accurate diagnosis is difficult and requires specialized dermatological testing. Limited treatment options are available but long-term prognosis is variable from patient to patient, based on individual case studies. Some skin symptoms recede with increasing age, while progressive neurological advancement of the disorder causes seizures and mental deterioration later in life for some patients.
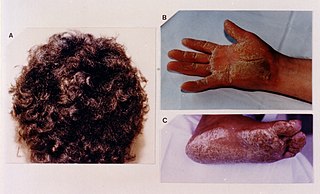
Woolly hair is a difficult to brush hair, usually present since birth and typically most severe in childhood. It has extreme curls and kinks and occurs in non-black people and is distinct from afro-textured hair. The hairs come together to form tight locks, unlike in afro-textured hair, where the hairs remain individual. Woolly hair can be generalised over the whole scalp, when it tends to run in families, or it may involve just part of the scalp as in woolly hair nevus.
Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I. It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome.
DOCK8 deficiency, also called DOCK8 immunodeficiency syndrome, is the autosomal recessive form of hyperimmunoglobulin E syndrome, a genetic disorder characterized by elevated immunoglobulin E levels, eosinophilia, and recurrent infections with staphylococcus and viruses. It is caused by a mutation in the DOCK8 gene.
Skin fragility-woolly hair-palmoplantar keratoderma syndrome is a very rare genetic disorder which is characterized by fragile skin which shows itself as blisters and erosion due to trauma that wouldn't typically cause those type of lesions, woolly hair with alopecia, nail dysplasia, widespread or local palmoplantar keratoderma associated with painful fissuring. Only 2 cases from two families have been described in medical literature.



