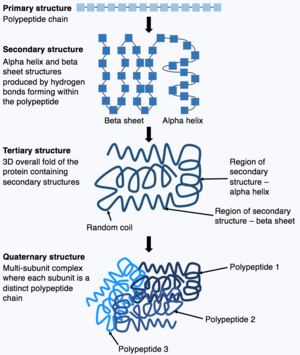
Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Protein tertiary structure is the three-dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains and the backbone may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure. A number of these structures may bind to each other, forming a quaternary structure.

Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
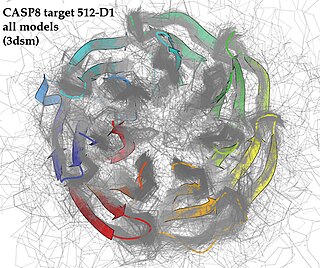
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.
Rosetta@home is a volunteer computing project researching protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker lab. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.

Sir Demis Hassabis is a British computer scientist, artificial intelligence researcher and entrepreneur. In his early career he was a video game AI programmer and designer, and an expert board games player. He is the chief executive officer and co-founder of DeepMind and Isomorphic Labs, and a UK Government AI Advisor.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
The global distance test (GDT), also written as GDT_TS to represent "total score", is a measure of similarity between two protein structures with known amino acid correspondences but different tertiary structures. It is most commonly used to compare the results of protein structure prediction to the experimentally determined structure as measured by X-ray crystallography, protein NMR, or, increasingly, cryoelectron microscopy. The metric was developed by Adam Zemla at Lawrence Livermore National Laboratory and originally implemented in the Local-Global Alignment (LGA) program. It is intended as a more accurate measurement than the common root-mean-square deviation (RMSD) metric - which is sensitive to outlier regions created, for example, by poor modeling of individual loop regions in a structure that is otherwise reasonably accurate. The conventional GDT_TS score is computed over the alpha carbon atoms and is reported as a percentage, ranging from 0 to 100. In general, the higher the GDT_TS score, the more closely a model approximates a given reference structure.

David Baker is an American biochemist and computational biologist who has pioneered methods to predict and design the three-dimensional structures of proteins. He is the Henrietta and Aubrey Davis Endowed Professor in Biochemistry and an adjunct professor of genome sciences, bioengineering, chemical engineering, computer science, and physics at the University of Washington. He serves as the director of the Rosetta Commons, a consortium of labs and researchers that develop biomolecular structure prediction and design software. The problem of protein structure prediction to which Baker has contributed significantly has now been largely solved by DeepMind using artificial intelligence. Baker is a Howard Hughes Medical Institute investigator and a member of the United States National Academy of Sciences. He is also the director of the University of Washington's Institute for Protein Design.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
Jianlin (Jack) Cheng is the William and Nancy Thompson Missouri Distinguished Professor in the Electrical Engineering and Computer Science (EECS) Department at the University of Missouri, Columbia. He earned his PhD from the University of California-Irvine in 2006, his MS degree from Utah State University in 2001, and his BS degree from Huazhong University of Science and Technology in 1994.

DeepMind Technologies Limited, doing business as Google DeepMind, is a British-American artificial intelligence research laboratory which serves as a subsidiary of Google. Founded in the UK in 2010, it was acquired by Google in 2014 and merged with Google AI's Google Brain division to become Google DeepMind in April 2023. The company is based in London, with research centres in Canada, France, Germany, and the United States.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
John Michael Jumper is an American senior research scientist at DeepMind Technologies. Jumper and his colleagues created AlphaFold, an artificial intelligence (AI) model to predict protein structures from their amino acid sequence with high accuracy. Jumper has stated that the AlphaFold team plans to release 100 million protein structures. The scientific journal Nature included Jumper as one of the ten "people who mattered" in science in their annual listing of Nature's 10 in 2021.
Nir1 or membrane-associated phosphatidylinositol transfer protein 3 (PITPNM3) is a mammalian protein that localizes to endoplasmic reticulum (ER) and plasma membrane (PM) membrane contact sites (MCS) and aids the transfer of phosphatidylinositol between these two membranes, potentially by recruiting additional proteins to the ER-PM MCS. It is encoded by the gene PITPNM3.

The Predicted Aligned Error (PAE) is a quantitative output produced by AlphaFold, a protein structure prediction system developed by DeepMind. PAE estimates the expected positional error for each residue in a predicted protein structure if it were aligned to a corresponding residue in the true protein structure. This measurement helps scientists assess the confidence in the relative positions and orientations of different parts of the predicted protein model.