Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.
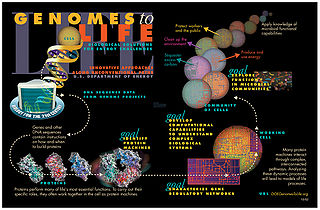
Systems biology is the computational and mathematical analysis and modeling of complex biological systems. It is a biology-based interdisciplinary field of study that focuses on complex interactions within biological systems, using a holistic approach to biological research.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
Modelling biological systems is a significant task of systems biology and mathematical biology. Computational systems biology aims to develop and use efficient algorithms, data structures, visualization and communication tools with the goal of computer modelling of biological systems. It involves the use of computer simulations of biological systems, including cellular subsystems, to both analyze and visualize the complex connections of these cellular processes.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.
Metabolic network modelling, also known as metabolic network reconstruction or metabolic pathway analysis, allows for an in-depth insight into the molecular mechanisms of a particular organism. In particular, these models correlate the genome with molecular physiology. A reconstruction breaks down metabolic pathways into their respective reactions and enzymes, and analyzes them within the perspective of the entire network. In simplified terms, a reconstruction collects all of the relevant metabolic information of an organism and compiles it in a mathematical model. Validation and analysis of reconstructions can allow identification of key features of metabolism such as growth yield, resource distribution, network robustness, and gene essentiality. This knowledge can then be applied to create novel biotechnology.

David Haussler is an American bioinformatician known for his work leading the team that assembled the first human genome sequence in the race to complete the Human Genome Project and subsequently for comparative genome analysis that deepens understanding the molecular function and evolution of the genome.

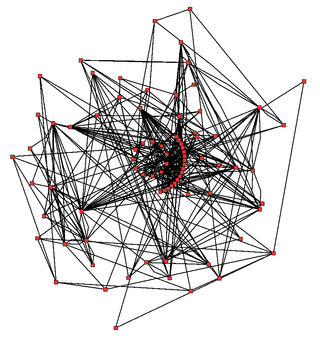
Biological network inference is the process of making inferences and predictions about biological networks. By using these networks to analyze patterns in biological systems, such as food-webs, we can visualize the nature and strength of these interactions between species, DNA, proteins, and more.
Physiomics is a systematic study of physiome in biology. Physiomics employs bioinformatics to construct networks of physiological features that are associated with genes, proteins and their networks. A few of the methods for determining individual relationships between the DNA sequence and physiological function include metabolic pathway engineering and RNAi analysis. The relationships derived from methods such as these are organized and processed computationally to form distinct networks. Computer models use these experimentally determined networks to develop further predictions of gene function.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Translational bioinformatics (TBI) is a field that emerged in the 2010s to study health informatics, focused on the convergence of molecular bioinformatics, biostatistics, statistical genetics and clinical informatics. Its focus is on applying informatics methodology to the increasing amount of biomedical and genomic data to formulate knowledge and medical tools, which can be utilized by scientists, clinicians, and patients. Furthermore, it involves applying biomedical research to improve human health through the use of computer-based information system. TBI employs data mining and analyzing biomedical informatics in order to generate clinical knowledge for application. Clinical knowledge includes finding similarities in patient populations, interpreting biological information to suggest therapy treatments and predict health outcomes.

Ron Shamir is an Israeli professor of computer science known for his work in graph theory and in computational biology. He holds the Raymond and Beverly Sackler Chair in Bioinformatics, and is the founder and former head of the Edmond J. Safra Center for Bioinformatics at Tel Aviv University.

Gary Stormo is an American geneticist and currently Joseph Erlanger Professor in the Department of Genetics and the Center for Genome Sciences and Systems Biology at Washington University School of Medicine in St Louis. He is considered one of the pioneers of bioinformatics and genomics. His research combines experimental and computational approaches in order to identify and predict regulatory sequences in DNA and RNA, and their contributions to the regulatory networks that control gene expression.
In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text mining derived associations including Mendelian, complex and environmental diseases.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
Christos A. Ouzounis is a computational biologist, a director of research at the CERTH, and Professor of Bioinformatics at Aristotle University in Thessaloniki.

Genome Informatics is a scientific study of information processing in genomes.