In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

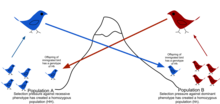
Natural selection is the differential survival and reproduction of individuals due to differences in phenotype. It is a key mechanism of evolution, the change in the heritable traits characteristic of a population over generations. Charles Darwin popularised the term "natural selection", contrasting it with artificial selection, which is intentional, whereas natural selection is not.
Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.

Motoo Kimura was a Japanese biologist best known for introducing the neutral theory of molecular evolution in 1968. He became one of the most influential theoretical population geneticists. He is remembered in genetics for his innovative use of diffusion equations to calculate the probability of fixation of beneficial, deleterious, or neutral alleles. Combining theoretical population genetics with molecular evolution data, he also developed the neutral theory of molecular evolution in which genetic drift is the main force changing allele frequencies. James F. Crow, himself a renowned population geneticist, considered Kimura to be one of the two greatest evolutionary geneticists, along with Gustave Malécot, after the great trio of the modern synthesis, Ronald Fisher, J. B. S. Haldane, and Sewall Wright.
In population genetics and population ecology, population size is a countable quantity representing the number of individual organisms in a population. Population size is directly associated with amount of genetic drift, and is the underlying cause of effects like population bottlenecks and the founder effect. Genetic drift is the major source of decrease of genetic diversity within populations which drives fixation and can potentially lead to speciation events.
Genetic load is the difference between the fitness of an average genotype in a population and the fitness of some reference genotype, which may be either the best present in a population, or may be the theoretically optimal genotype. The average individual taken from a population with a low genetic load will generally, when grown in the same conditions, have more surviving offspring than the average individual from a population with a high genetic load. Genetic load can also be seen as reduced fitness at the population level compared to what the population would have if all individuals had the reference high-fitness genotype. High genetic load may put a population in danger of extinction.
Mutation–selection balance is an equilibrium in the number of deleterious alleles in a population that occurs when the rate at which deleterious alleles are created by mutation equals the rate at which deleterious alleles are eliminated by selection. The majority of genetic mutations are neutral or deleterious; beneficial mutations are relatively rare. The resulting influx of deleterious mutations into a population over time is counteracted by negative selection, which acts to purge deleterious mutations. Setting aside other factors, the equilibrium number of deleterious alleles is then determined by a balance between the deleterious mutation rate and the rate at which selection purges those mutations.
Genetic hitchhiking, also called genetic draft or the hitchhiking effect, is when an allele changes frequency not because it itself is under natural selection, but because it is near another gene that is undergoing a selective sweep and that is on the same DNA chain. When one gene goes through a selective sweep, any other nearby polymorphisms that are in linkage disequilibrium will tend to change their allele frequencies too. Selective sweeps happen when newly appeared mutations are advantageous and increase in frequency. Neutral or even slightly deleterious alleles that happen to be close by on the chromosome 'hitchhike' along with the sweep. In contrast, effects on a neutral locus due to linkage disequilibrium with newly appeared deleterious mutations are called background selection. Both genetic hitchhiking and background selection are stochastic (random) evolutionary forces, like genetic drift.

The Neutral Theory of Molecular Evolution is an influential monograph written in 1983 by Japanese evolutionary biologist Motoo Kimura. While the neutral theory of molecular evolution existed since his article in 1968, Kimura felt the need to write a monograph with up-to-date information and evidences showing the importance of his theory in evolution.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will be lost or will replace all other alleles of the gene. That loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.
In natural selection, negative selection or purifying selection is the selective removal of alleles that are deleterious. This can result in stabilising selection through the purging of deleterious genetic polymorphisms that arise through random mutations.
In population genetics, fixation is the change in a gene pool from a situation where there exists at least two variants of a particular gene (allele) in a given population to a situation where only one of the alleles remains. That is, the allele becomes fixed. In the absence of mutation or heterozygote advantage, any allele must eventually either be lost completely from the population, or fixed, i.e. permanently established at 100% frequency in the population. Whether a gene will ultimately be lost or fixed is dependent on selection coefficients and chance fluctuations in allelic proportions. Fixation can refer to a gene in general or particular nucleotide position in the DNA chain (locus).
The nearly neutral theory of molecular evolution is a modification of the neutral theory of molecular evolution that accounts for the fact that not all mutations are either so deleterious such that they can be ignored, or else neutral. Slightly deleterious mutations are reliably purged only when their selection coefficient are greater than one divided by the effective population size. In larger populations, a higher proportion of mutations exceed this threshold for which genetic drift cannot overpower selection, leading to fewer fixation events and so slower molecular evolution.
The history of molecular evolution starts in the early 20th century with "comparative biochemistry", but the field of molecular evolution came into its own in the 1960s and 1970s, following the rise of molecular biology. The advent of protein sequencing allowed molecular biologists to create phylogenies based on sequence comparison, and to use the differences between homologous sequences as a molecular clock to estimate the time since the last common ancestor. In the late 1960s, the neutral theory of molecular evolution provided a theoretical basis for the molecular clock, though both the clock and the neutral theory were controversial, since most evolutionary biologists held strongly to panselectionism, with natural selection as the only important cause of evolutionary change. After the 1970s, nucleic acid sequencing allowed molecular evolution to reach beyond proteins to highly conserved ribosomal RNA sequences, the foundation of a reconceptualization of the early history of life.

The Bateson–Dobzhansky–Muller model, also known as Dobzhansky–Muller model, is a model of the evolution of genetic incompatibility, important in understanding the evolution of reproductive isolation during speciation and the role of natural selection in bringing it about. The theory was first described by William Bateson in 1909, then independently described by Theodosius Dobzhansky in 1934, and later elaborated in different forms by Herman Muller, H. Allen Orr and Sergey Gavrilets.
Genetic purging is the increased pressure of natural selection against deleterious alleles prompted by inbreeding.

Epistasis is a phenomenon in genetics in which the effect of a gene mutation is dependent on the presence or absence of mutations in one or more other genes, respectively termed modifier genes. In other words, the effect of the mutation is dependent on the genetic background in which it appears. Epistatic mutations therefore have different effects on their own than when they occur together. Originally, the term epistasis specifically meant that the effect of a gene variant is masked by that of different gene.
Bias in the introduction of variation is a theory in the domain of evolutionary biology that asserts biases in the introduction of heritable variation are reflected in the outcome of evolution. It is relevant to topics in molecular evolution, evo-devo, and self-organization. In the context of this theory, "introduction" ("origination") is a technical term for events that shift an allele frequency upward from zero. Formal models demonstrate that when an evolutionary process depends on introduction events, mutational and developmental biases in the generation of variation may influence the course of evolution by a first come, first served effect, so that evolution reflects the arrival of the likelier, not just the survival of the fitter. Whereas mutational explanations for evolutionary patterns are typically assumed to imply or require neutral evolution, the theory of arrival biases distinctively predicts the possibility of mutation-biased adaptation. Direct evidence for the theory comes from laboratory studies showing that adaptive changes are systematically enriched for mutationally likely types of changes. Retrospective analyses of natural cases of adaptation also provide support for the theory. This theory is notable as an example of contemporary structuralist thinking, contrasting with a classical functionalist view in which the course of evolution is determined by natural selection.