Evolution is the change in the heritable characteristics of biological populations over successive generations. It occurs when evolutionary processes such as natural selection and genetic drift act on genetic variation, resulting in certain characteristics becoming more or less common within a population over successive generations. The process of evolution has given rise to biodiversity at every level of biological organisation.
Microevolution is the change in allele frequencies that occurs over time within a population. This change is due to four different processes: mutation, selection, gene flow and genetic drift. This change happens over a relatively short amount of time compared to the changes termed macroevolution.

In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
Junk DNA is a DNA sequence that has no relevant biological function. Most organisms have some junk DNA in their genomes—mostly pseudogenes and fragments of transposons and viruses—but it is possible that some organisms have substantial amounts of junk DNA.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.
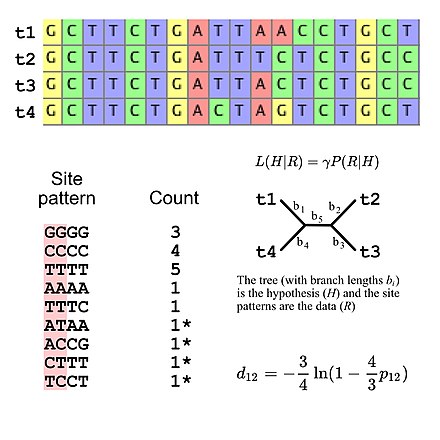
The molecular clock is a figurative term for a technique that uses the mutation rate of biomolecules to deduce the time in prehistory when two or more life forms diverged. The biomolecular data used for such calculations are usually nucleotide sequences for DNA, RNA, or amino acid sequences for proteins.
Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
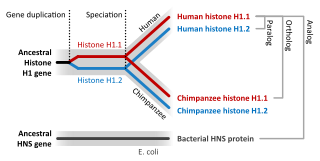
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.
In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will be lost or will replace all other alleles of the gene. That loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.

Masatoshi Nei was a Japanese-born American evolutionary biologist.
The history of molecular evolution starts in the early 20th century with "comparative biochemistry", but the field of molecular evolution came into its own in the 1960s and 1970s, following the rise of molecular biology. The advent of protein sequencing allowed molecular biologists to create phylogenies based on sequence comparison, and to use the differences between homologous sequences as a molecular clock to estimate the time since the last common ancestor. In the late 1960s, the neutral theory of molecular evolution provided a theoretical basis for the molecular clock, though both the clock and the neutral theory were controversial, since most evolutionary biologists held strongly to panselectionism, with natural selection as the only important cause of evolutionary change. After the 1970s, nucleic acid sequencing allowed molecular evolution to reach beyond proteins to highly conserved ribosomal RNA sequences, the foundation of a reconceptualization of the early history of life.

Genome evolution is the process by which a genome changes in structure (sequence) or size over time. The study of genome evolution involves multiple fields such as structural analysis of the genome, the study of genomic parasites, gene and ancient genome duplications, polyploidy, and comparative genomics. Genome evolution is a constantly changing and evolving field due to the steadily growing number of sequenced genomes, both prokaryotic and eukaryotic, available to the scientific community and the public at large.
An overlapping gene is a gene whose expressible nucleotide sequence partially overlaps with the expressible nucleotide sequence of another gene. In this way, a nucleotide sequence may make a contribution to the function of one or more gene products. Overlapping genes are present in and a fundamental feature of both cellular and viral genomes. The current definition of an overlapping gene varies significantly between eukaryotes, prokaryotes, and viruses. In prokaryotes and viruses overlap must be between coding sequences but not mRNA transcripts, and is defined when these coding sequences share a nucleotide on either the same or opposite strands. In eukaryotes, gene overlap is almost always defined as mRNA transcript overlap. Specifically, a gene overlap in eukaryotes is defined when at least one nucleotide is shared between the boundaries of the primary mRNA transcripts of two or more genes, such that a DNA base mutation at any point of the overlapping region would affect the transcripts of all genes involved. This definition includes 5′ and 3′ untranslated regions (UTRs) along with introns.

De novo gene birth is the process by which new genes evolve from non-coding DNA. De novo genes represent a subset of novel genes, and may be protein-coding or instead act as RNA genes. The processes that govern de novo gene birth are not well understood, although several models exist that describe possible mechanisms by which de novo gene birth may occur.