Related Research Articles

Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.

Hemoglobinopathy is the medical term for a group of inherited blood disorders involving the hemoglobin, the protein of red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen. This can be due to a lower than normal number of red blood cells, a reduction in the amount of hemoglobin available for oxygen transport, or abnormalities in hemoglobin that impair its function. The name is derived from Ancient Greek ἀν- (an-) 'not', and αἷμα (haima) 'blood'. When anemia comes on slowly, the symptoms are often vague, such as tiredness, weakness, shortness of breath, headaches, and a reduced ability to exercise. When anemia is acute, symptoms may include confusion, feeling like one is going to pass out, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Additional symptoms may occur depending on the underlying cause. Anemia can be temporary or long term and can range from mild to severe.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time. Thalassemia is also known as Cooley's anemia or Mediterranean anemia.

Kernicterus is a bilirubin-induced brain dysfunction. The term was coined in 1904 by Christian Georg Schmorl. Bilirubin is a naturally occurring substance in the body of humans and many other animals, but it is neurotoxic when its concentration in the blood is too high, a condition known as hyperbilirubinemia. Hyperbilirubinemia may cause bilirubin to accumulate in the grey matter of the central nervous system, potentially causing irreversible neurological damage. Depending on the level of exposure, the effects range from clinically unnoticeable to severe brain damage and even death.
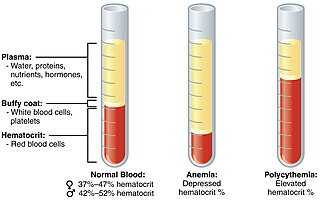
Polycythemia is a laboratory finding in which the hematocrit and/or hemoglobin concentration are increased in the blood. Polycythemia is sometimes called erythrocytosis, and there is significant overlap in the two findings, but the terms are not the same: polycythemia describes any increase in hematocrit and/or hemoglobin, while erythrocytosis describes an increase specifically in the number of red blood cells in the blood.

Hemolytic disease of the newborn, also known as hemolytic disease of the fetus and newborn, HDN, HDFN, or erythroblastosis fetalis, is an alloimmune condition that develops in a fetus at or around birth, when the IgG molecules produced by the mother pass through the placenta. Among these antibodies are some which attack antigens on the red blood cells in the fetal circulation, breaking down and destroying the cells. The fetus can develop reticulocytosis and anemia. The intensity of this fetal disease ranges from mild to very severe, and fetal death from heart failure can occur. When the disease is moderate or severe, many erythroblasts are present in the fetal blood, earning these forms of the disease the name erythroblastosis fetalis.
Autoimmune hemolytic anemia (AIHA) occurs when antibodies directed against the person's own red blood cells (RBCs) cause them to burst (lyse), leading to an insufficient number of oxygen-carrying red blood cells in the circulation. The lifetime of the RBCs is reduced from the normal 100–120 days to just a few days in serious cases. The intracellular components of the RBCs are released into the circulating blood and into tissues, leading to some of the characteristic symptoms of this condition. The antibodies are usually directed against high-incidence antigens, therefore they also commonly act on allogenic RBCs. AIHA is a relatively rare condition, with an incidence of 5–10 cases per 1 million persons per year in the warm-antibody type and 0.45 to 1.9 cases per 1 million persons per year in the cold antibody type. Autoimmune hemolysis might be a precursor of later onset systemic lupus erythematosus.
Sickle cell trait describes a condition in which a person has one abnormal allele of the hemoglobin beta gene, but does not display the severe symptoms of sickle cell disease that occur in a person who has two copies of that allele. Those who are heterozygous for the sickle cell allele produce both normal and abnormal hemoglobin.
Reticulocytopenia is the medical term for an abnormal decrease in circulating red blood cell precursors (reticulocytes) that can lead to anemia due to resulting low red blood cell (erythrocyte) production. Reticulocytopenia may be an isolated finding or it may not be associated with abnormalities in other hematopoietic cell lineages such as those that produce white blood cells (leukocytes) or platelets (thrombocytes), a decrease in all three of these lineages is referred to as pancytopenia.
Hemolytic disease of the newborn (anti-Rhc) can range from a mild to a severe disease. It is the third most common cause of severe HDN. Rh disease is the most common and hemolytic disease of the newborn (anti-Kell) is the second most common cause of severe HDN. It occurs more commonly in women who are Rh D negative.
Hemolytic disease of the newborn (anti-RhE) is caused by the anti-RhE antibody of the Rh blood group system. The anti-RhE antibody can be naturally occurring, or arise following immune sensitization after a blood transfusion or pregnancy.
Hematologic diseases are disorders which primarily affect the blood and blood-forming organs. Hematologic diseases include rare genetic disorders, anemia, HIV, sickle cell disease and complications from chemotherapy or transfusions.

Sickle cell disease (SCD), also simply called sickle cell, is a group of hemoglobin-related blood disorders typically inherited. The most common type is known as sickle cell anemia. It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells. This leads to a rigid, sickle-like shape under certain circumstances. Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain in joints, anemia, swelling in the hands and feet, bacterial infections, dizziness and stroke. Long-term pain may develop as people get older. The average life expectancy in the developed world is 40 to 60 years. It often gets worse within age. All the major organs are affected by sickle cell disease. The liver, heart, kidneys, gallbladder, eyes, bones, and joints also can suffer damage from the abnormal functions of the sickle cells, and their inability to flow through the small blood vessels correctly.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.

Blueberry muffin baby, also known as extramedullary hematopoiesis, describes a newborn baby with multiple purpura, associated with several non-cancerous and cancerous conditions in which extra blood is produced in the skin. The bumps range from one to seven mm, do not blanch and have a tendency to occur on the head, neck and trunk. They often fade by three to six weeks after birth, leaving brownish marks. When due to a cancer, the bumps tend to be fewer, firmer and larger.
This page is currently under construction.
Red blood cells (erythrocytes) from donors contain normal hemoglobin (HbA), and transfusion of normal red blood cells into people with sickle cell disease reduces the percentage of red cells in the circulation containing the abnormal hemoglobin (HbS). Although transfusion of donor red blood cells can ameliorate and even prevent complications of sickle cell disease in certain circumstances, transfusion therapy is not universally beneficial in sickle cell disease.
Hemoglobin H disease, also called alpha-thalassemia intermedia, is a disease affecting hemoglobin, the oxygen carrying molecule within red blood cells. It is a form of Alpha-thalassemia which most commonly occurs due to deletion of 3 out of 4 of the α-globin genes.
Hemolytic jaundice, also known as prehepatic jaundice, is a type of jaundice arising from hemolysis or excessive destruction of red blood cells, when the byproduct bilirubin is not excreted by the hepatic cells quickly enough. Unless the patient is concurrently affected by hepatic dysfunctions or is experiencing hepatocellular damage, the liver does not contribute to this type of jaundice.
References
- 1 2 3 4 "Exchange transfusion". Nlm.nih.gov. MedlinePlus.
- ↑ "Spectra Optia for automatic red blood cell exchange in patients with sickle cell disease | Guidance and guidelines | NICE". www.nice.org.uk. 2 March 2016. Retrieved 2019-01-04.
- 1 2 3 4 5 Davis, Bernard A.; Allard, Shubha; Qureshi, Amrana; Porter, John B.; Pancham, Shivan; Win, Nay; Cho, Gavin; Ryan, Kate (2017). "Guidelines on red cell transfusion in sickle cell disease. Part I: principles and laboratory aspects". British Journal of Haematology. 176 (2): 179–191. doi: 10.1111/bjh.14346 . ISSN 1365-2141. PMID 28092109. S2CID 3462324.
- 1 2 3 4 5 6 "Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014 | National Heart, Lung, and Blood Institute (NHLBI)". www.nhlbi.nih.gov. Retrieved 2019-01-04.
- 1 2 3 4 5 6 "Manual exchange blood transfusion protocol". www.gosh.nhs.uk. Archived from the original on 2019-04-12. Retrieved 2019-01-04.
- 1 2 3 4 5 6 7 Davis, Bernard A.; Allard, Shubha; Qureshi, Amrana; Porter, John B.; Pancham, Shivan; Win, Nay; Cho, Gavin; Ryan, Kate (2017). "Guidelines on red cell transfusion in sickle cell disease Part II: indications for transfusion" (PDF). British Journal of Haematology. 176 (2): 192–209. doi:10.1111/bjh.14383. ISSN 1365-2141. PMID 27858994. S2CID 3534824.
- ↑ Estcourt, Lise J; Fortin, Patricia M; Trivella, Marialena; Hopewell, Sally (2016-04-06). "Preoperative blood transfusions for sickle cell disease". Cochrane Database of Systematic Reviews. 4 (4): CD003149. doi:10.1002/14651858.cd003149.pub3. ISSN 1465-1858. PMC 4854326 . PMID 27049331.
- 1 2 3 Özek, Eren; Soll, Roger; Schimmel, Michael S (2010-01-20). "Partial exchange transfusion to prevent neurodevelopmental disability in infants with polycythemia". Cochrane Database of Systematic Reviews (1): CD005089. doi:10.1002/14651858.cd005089.pub2. ISSN 1465-1858. PMID 20091569.
- ↑ Paul, Vinod K.; Deorari, Ashok; Agarwal, Ramesh; Sankar, M. Jeeva (2010-10-01). "Management of Polycythemia in Neonates". The Indian Journal of Pediatrics. 77 (10): 1117–1121. doi:10.1007/s12098-010-0177-z. ISSN 0973-7693. PMID 20725868. S2CID 20174315.
- ↑ Bada, H. S.; Korones, S. B.; Kolni, H. W.; Fitch, C. W.; Ford, D. L.; Magill, H. L.; Anderson, G. D.; Wong, S. P. (1986). "Partial plasma exchange transfusion improves cerebral hemodynamics in symptomatic neonatal polycythemia". The American Journal of the Medical Sciences. 291 (3): 157–163. doi:10.1097/00000441-198603000-00003. ISSN 0002-9629. PMID 3953635. S2CID 35878878.
- ↑ Dongare, H. C.; Khatib, K. I. (2016). "JCDR - Exchange blood transfusion, Malaria, Plasmodium falciparum". Journal of Clinical and Diagnostic Research. 10 (2): OD05–6. doi:10.7860/jcdr/2016/16341.7190. PMC 4800569 . PMID 27042503.
- 1 2 3 4 "CDC - Malaria - Exchange Transfusion for Treatment of Severe Malaria No Longer Recommended". www.cdc.gov. United States Centers for Disease Control and Prevention. 28 June 2017. Retrieved 2019-01-04.
- ↑ "Scheduled Outpatient Red Blood Cell Exchange Program Reduces Admission and Complications in Sickle Cell Disease". ashpublications.org.
- ↑ "Alexander Wiener biography". Bbguy.org. Archived from the original on 2014-08-12. Retrieved 2014-07-28.