
Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Density-functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals, i.e. functions of another function. In the case of DFT, these are functionals of the spatially dependent electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.
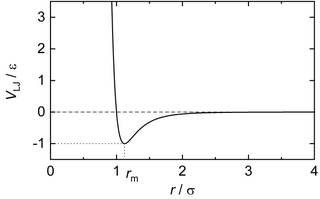
In computational chemistry, the Lennard-Jones potential is an intermolecular pair potential. Out of all the intermolecular potentials, the Lennard-Jones potential is probably the one that has been the most extensively studied. It is considered an archetype model for simple yet realistic intermolecular interactions.

In theoretical physics, the pilot wave theory, also known as Bohmian mechanics, was the first known example of a hidden-variable theory, presented by Louis de Broglie in 1927. Its more modern version, the de Broglie–Bohm theory, interprets quantum mechanics as a deterministic theory, avoiding troublesome notions such as wave–particle duality, instantaneous wave function collapse, and the paradox of Schrödinger's cat. To solve these problems, the theory is inherently nonlocal.
In probability theory and mathematical physics, a random matrix is a matrix-valued random variable—that is, a matrix in which some or all elements are random variables. Many important properties of physical systems can be represented mathematically as matrix problems. For example, the thermal conductivity of a lattice can be computed from the dynamical matrix of the particle-particle interactions within the lattice.

In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.
Implicit solvation is a method to represent solvent as a continuous medium instead of individual “explicit” solvent molecules, most often used in molecular dynamics simulations and in other applications of molecular mechanics. The method is often applied to estimate free energy of solute-solvent interactions in structural and chemical processes, such as folding or conformational transitions of proteins, DNA, RNA, and polysaccharides, association of biological macromolecules with ligands, or transport of drugs across biological membranes.

In computational chemistry, a water model is used to simulate and thermodynamically calculate water clusters, liquid water, and aqueous solutions with explicit solvent. The models are determined from quantum mechanics, molecular mechanics, experimental results, and these combinations. To imitate a specific nature of molecules, many types of models have been developed. In general, these can be classified by the following three points; (i) the number of interaction points called site, (ii) whether the model is rigid or flexible, (iii) whether the model includes polarization effects.
In physics, a pair potential is a function that describes the potential energy of two interacting objects solely as a function of the distance between them.
The Berendsen thermostat is an algorithm to re-scale the velocities of particles in molecular dynamics simulations to control the simulation temperature.
The Wang and Landau algorithm, proposed by Fugao Wang and David P. Landau, is a Monte Carlo method designed to estimate the density of states of a system. The method performs a non-Markovian random walk to build the density of states by quickly visiting all the available energy spectrum. The Wang and Landau algorithm is an important method to obtain the density of states required to perform a multicanonical simulation.

CP2K is a freely available (GPL) quantum chemistry and solid state physics program package, written in Fortran 2008, to perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. It provides a general framework for different methods: density functional theory (DFT) using a mixed Gaussian and plane waves approach (GPW) via LDA, GGA, MP2, or RPA levels of theory, classical pair and many-body potentials, semi-empirical and tight-binding Hamiltonians, as well as Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid schemes relying on the Gaussian Expansion of the Electrostatic Potential (GEEP). The Gaussian and Augmented Plane Waves method (GAPW) as an extension of the GPW method allows for all-electron calculations. CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method.
Crystal structure prediction (CSP) is the calculation of the crystal structures of solids from first principles. Reliable methods of predicting the crystal structure of a compound, based only on its composition, has been a goal of the physical sciences since the 1950s. Computational methods employed include simulated annealing, evolutionary algorithms, distributed multipole analysis, random sampling, basin-hopping, data mining, density functional theory and molecular mechanics.
Multi-particle collision dynamics (MPC), also known as stochastic rotation dynamics (SRD), is a particle-based mesoscale simulation technique for complex fluids which fully incorporates thermal fluctuations and hydrodynamic interactions. Coupling of embedded particles to the coarse-grained solvent is achieved through molecular dynamics.
Local elevation is a technique used in computational chemistry or physics, mainly in the field of molecular simulation. It was developed in 1994 by Huber, Torda and van Gunsteren to enhance the searching of conformational space in molecular dynamics simulations and is available in the GROMOS software for molecular dynamics simulation. The method was, together with the conformational flooding method, the first to introduce memory dependence into molecular simulations. Many recent methods build on the principles of the local elevation technique, including the Engkvist-Karlström, adaptive biasing force, Wang–Landau, metadynamics, adaptively biased molecular dynamics, adaptive reaction coordinate forces, and local elevation umbrella sampling methods. The basic principle of the method is to add a memory-dependent potential energy term in the simulation so as to prevent the simulation to revisit already sampled configurations, which leads to the increased probability of discovering new configurations. The method can be seen as a continuous variant of the Tabu search method.

Interatomic potentials are mathematical functions to calculate the potential energy of a system of atoms with given positions in space. Interatomic potentials are widely used as the physical basis of molecular mechanics and molecular dynamics simulations in computational chemistry, computational physics and computational materials science to explain and predict materials properties. Examples of quantitative properties and qualitative phenomena that are explored with interatomic potentials include lattice parameters, surface energies, interfacial energies, adsorption, cohesion, thermal expansion, and elastic and plastic material behavior, as well as chemical reactions.
Boson sampling is a restricted model of non-universal quantum computation introduced by Scott Aaronson and Alex Arkhipov after the original work of Lidror Troyansky and Naftali Tishby, that explored possible usage of boson scattering to evaluate expectation values of permanents of matrices. The model consists of sampling from the probability distribution of identical bosons scattered by a linear interferometer. Although the problem is well defined for any bosonic particles, its photonic version is currently considered as the most promising platform for a scalable implementation of a boson sampling device, which makes it a non-universal approach to linear optical quantum computing. Moreover, while not universal, the boson sampling scheme is strongly believed to implement computing tasks which are hard to implement with classical computers by using far fewer physical resources than a full linear-optical quantum computing setup. This advantage makes it an ideal candidate for demonstrating the power of quantum computation in the near term.
PLUMED is an open-source library implementing enhanced-sampling algorithms, various free-energy methods, and analysis tools for molecular dynamics simulations. It is designed to be used together with ACEMD, AMBER, DL_POLY, GROMACS, LAMMPS, NAMD, OpenMM, ABIN, CP2K, i-PI, PINY-MD, and Quantum ESPRESSO, but it can also be used to together with analysis and visualization tools VMD, HTMD, and OpenPathSampling.
In physics and chemistry, photoemission orbital tomography is a combined experimental / theoretical approach which reveals information about the spatial distribution of individual molecular orbitals. Experimentally, it uses angle-resolved photoemission spectroscopy (ARPES) to obtain constant binding energy photoemission angular distribution maps, so-called tomograms, to reveal information about the electron probability distribution in molecular orbitals. Theoretically, one rationalizes these tomograms as hemispherical cuts through the molecular orbital in momentum space. This interpretation relies on the assumption of a plane wave final state, i.e., the idea that the outgoing electron can be treated as a free electron, which can be further exploited to reconstruct real-space images of molecular orbitals on a sub-Ångström length scale in two or three dimensions. Presently, POT has been applied to various organic molecules forming well-oriented monolayers on single crystal surfaces or to two-dimensional materials.







































