Force spectroscopy is a set of techniques for the study of the interactions and the binding forces between individual molecules. These methods can be used to measure the mechanical properties of single polymer molecules or proteins, or individual chemical bonds. The name "force spectroscopy", although widely used in the scientific community, is somewhat misleading, because there is no true matter-radiation interaction.
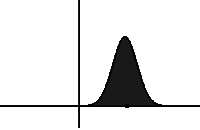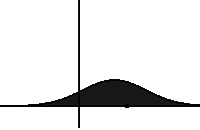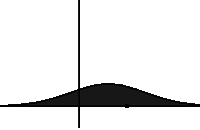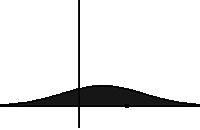
In statistical mechanics and information theory, the Fokker–Planck equation is a partial differential equation that describes the time evolution of the probability density function of the velocity of a particle under the influence of drag forces and random forces, as in Brownian motion. The equation can be generalized to other observables as well. The Fokker-Planck equation has multiple applications in information theory, graph theory, data science, finance, economics etc.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Chemistry at Harvard Macromolecular Mechanics (CHARMM) is the name of a widely used set of force fields for molecular dynamics, and the name for the molecular dynamics simulation and analysis computer software package associated with them. The CHARMM Development Project involves a worldwide network of developers working with Martin Karplus and his group at Harvard to develop and maintain the CHARMM program. Licenses for this software are available, for a fee, to people and groups working in academia.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.

Smoothed-particle hydrodynamics (SPH) is a computational method used for simulating the mechanics of continuum media, such as solid mechanics and fluid flows. It was developed by Gingold and Monaghan and Lucy in 1977, initially for astrophysical problems. It has been used in many fields of research, including astrophysics, ballistics, volcanology, and oceanography. It is a meshfree Lagrangian method, and the resolution of the method can easily be adjusted with respect to variables such as density.

In nuclear and materials physics, stopping power is the retarding force acting on charged particles, typically alpha and beta particles, due to interaction with matter, resulting in loss of particle kinetic energy. Its application is important in areas such as radiation protection, ion implantation and nuclear medicine.
In physics, Langevin dynamics is an approach to the mathematical modeling of the dynamics of molecular systems. It was originally developed by French physicist Paul Langevin. The approach is characterized by the use of simplified models while accounting for omitted degrees of freedom by the use of stochastic differential equations. Langevin dynamics simulations are a kind of Monte Carlo simulation.
In physics, Brownian dynamics is a mathematical approach for describing the dynamics of molecular systems in the diffusive regime. It is a simplified version of Langevin dynamics and corresponds to the limit where no average acceleration takes place. This approximation is also known as overdamped Langevin dynamics or as Langevin dynamics without inertia.
In atomic, molecular, and optical physics and quantum chemistry, the molecular Hamiltonian is the Hamiltonian operator representing the energy of the electrons and nuclei in a molecule. This operator and the associated Schrödinger equation play a central role in computational chemistry and physics for computing properties of molecules and aggregates of molecules, such as thermal conductivity, specific heat, electrical conductivity, optical, and magnetic properties, and reactivity.

In protein structure prediction, statistical potentials or knowledge-based potentials are scoring functions derived from an analysis of known protein structures in the Protein Data Bank (PDB).
Implicit solvation is a method to represent solvent as a continuous medium instead of individual “explicit” solvent molecules, most often used in molecular dynamics simulations and in other applications of molecular mechanics. The method is often applied to estimate free energy of solute-solvent interactions in structural and chemical processes, such as folding or conformational transitions of proteins, DNA, RNA, and polysaccharides, association of biological macromolecules with ligands, or transport of drugs across biological membranes.
The quantum potential or quantum potentiality is a central concept of the de Broglie–Bohm formulation of quantum mechanics, introduced by David Bohm in 1952.
In computational chemistry, a constraint algorithm is a method for satisfying the Newtonian motion of a rigid body which consists of mass points. A restraint algorithm is used to ensure that the distance between mass points is maintained. The general steps involved are: (i) choose novel unconstrained coordinates, (ii) introduce explicit constraint forces, (iii) minimize constraint forces implicitly by the technique of Lagrange multipliers or projection methods.
Free energy perturbation (FEP) is a method based on statistical mechanics that is used in computational chemistry for computing free energy differences from molecular dynamics or Metropolis Monte Carlo simulations.

Umbrella sampling is a technique in computational physics and chemistry, used to improve sampling of a system where ergodicity is hindered by the form of the system's energy landscape. It was first suggested by Torrie and Valleau in 1977. It is a particular physical application of the more general importance sampling in statistics.

In physics, Lagrangian mechanics is a formulation of classical mechanics founded on the stationary-action principle. It was introduced by the Italian-French mathematician and astronomer Joseph-Louis Lagrange in his 1788 work, Mécanique analytique.
The Kirkwood–Buff (KB) solution theory, due to John G. Kirkwood and Frank P. Buff, links macroscopic (bulk) properties to microscopic (molecular) details. Using statistical mechanics, the KB theory derives thermodynamic quantities from pair correlation functions between all molecules in a multi-component solution. The KB theory proves to be a valuable tool for validation of molecular simulations, as well as for the molecular-resolution elucidation of the mechanisms underlying various physical processes. For example, it has numerous applications in biologically relevant systems.
In computational chemistry, a solvent model is a computational method that accounts for the behavior of solvated condensed phases. Solvent models enable simulations and thermodynamic calculations applicable to reactions and processes which take place in solution. These include biological, chemical and environmental processes. Such calculations can lead to new predictions about the physical processes occurring by improved understanding.








