Electrophysiology is the branch of physiology that studies the electrical properties of biological cells and tissues. It involves measurements of voltage changes or electric current or manipulations on a wide variety of scales from single ion channel proteins to whole organs like the heart. In neuroscience, it includes measurements of the electrical activity of neurons, and, in particular, action potential activity. Recordings of large-scale electric signals from the nervous system, such as electroencephalography, may also be referred to as electrophysiological recordings. They are useful for electrodiagnosis and monitoring.
A cell type is a classification used to identify cells that share morphological or phenotypical features. A multicellular organism may contain cells of a number of widely differing and specialized cell types, such as muscle cells and skin cells, that differ both in appearance and function yet have identical genomic sequences. Cells may have the same genotype, but belong to different cell types due to the differential regulation of the genes they contain. Classification of a specific cell type is often done through the use of microscopy. Recent developments in single cell RNA sequencing facilitated classification of cell types based on shared gene expression patterns. This has led to the discovery of many new cell types in e.g. mouse cortex, hippocampus, dorsal root ganglion and spinal cord.

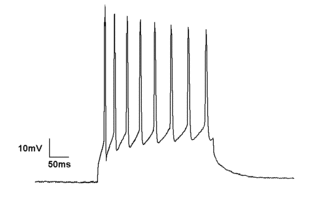
The patch clamp technique is a laboratory technique in electrophysiology used to study ionic currents in individual isolated living cells, tissue sections, or patches of cell membrane. The technique is especially useful in the study of excitable cells such as neurons, cardiomyocytes, muscle fibers, and pancreatic beta cells, and can also be applied to the study of bacterial ion channels in specially prepared giant spheroplasts.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.
Microelectrode arrays (MEAs) are devices that contain multiple microelectrodes through which neural signals are obtained or delivered, essentially serving as neural interfaces that connect neurons to electronic circuitry. There are two general classes of MEAs: implantable MEAs, used in vivo, and non-implantable MEAs, used in vitro.

RNA-Seq is a sequencing technique that uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample, representing an aggregated snapshot of the cells' dynamic pool of RNAs, also known as transcriptome.
Automated patch clamping is beginning to replace manual patch clamping as a method to measure the electrical activity of individual cells. Different techniques are used to automate patch clamp recordings from cells in cell culture and in vivo. This work has been ongoing since the late 1990s by research labs and companies trying to reduce its complexity and cost of patch clamping manually. Patch clamping for a long time was considered an art form and is still very time consuming and tedious, especially in vivo. The automation techniques try to reduce user error and variability in obtaining quality electrophysiology recordings from single cells.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.

A transcriptome in vivo analysis tag is a multifunctional, photoactivatable mRNA-capture molecule designed for isolating mRNA from a single cell in complex tissues.
Cajal–Retzius cells are a heterogeneous population of morphologically and molecularly distinct reelin-producing cell types in the marginal zone/layer I of the developmental cerebral cortex and in the immature hippocampus of different species and at different times during embryogenesis and postnatal life.
Frank Werblin is Professor of the Graduate School, Division of Neurobiology at the University of California, Berkeley.
Single-cell transcriptomics examines the gene expression level of individual cells in a given population by simultaneously measuring the RNA concentration of hundreds to thousands of genes. Single-cell transcriptomics makes it possible to unravel heterogeneous cell populations, reconstruct cellular developmental pathways, and model transcriptional dynamics — all previously masked in bulk RNA sequencing.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
MAPseq or Multiplexed Analysis of Projections by Sequencing is a RNA-Seq based method for high-throughput mapping of neuronal projections. It was developed by Anthony M. Zador and his team at Cold Spring Harbor Laboratory and published in Neuron, a Cell Press magazine.

Spatial transcriptomics is a method for assigning cell types to their locations in the histological sections and can also be used to determine subcellular localization of mRNA molecules. First described in 2016 by Ståhl et al., it has since undergone a variety of improvements and modifications.
CITE-Seq is a method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level. So far, the method has been demonstrated to work with only a few proteins per cell. As such, it provides an additional layer of information for the same cell by combining both proteomics and transcriptomics data. For phenotyping, this method has been shown to be as accurate as flow cytometry by the groups that developed it. It is currently one of the main methods, along with REAP-Seq, to evaluate both gene expression and protein levels simultaneously in different species.
snRNA-seq, also known as single nucleus RNA sequencing, single nuclei RNA sequencing or sNuc-seq, is an RNA sequencing method for profiling gene expression in cells which are difficult to isolate, such as those from tissues that are archived or which are hard to be dissociated. It is an alternative to single cell RNA seq (scRNA-seq), as it analyzes nuclei instead of intact cells.
Deterministic Barcoding in Tissue for Spatial Omics Sequencing (DBiT-seq) was developed at Yale University by Rong Fan and colleagues in 2020 to create a multi-omics approach for studying spatial gene expression heterogenicity within a tissue sample. This method can used for the co-mapping mRNA and protein levels at a near single-cell resolution in fresh or frozen formaldehyde-fixed tissue samples. DBiT-seq utilizes next generation sequencing (NGS) and microfluidics. This method allows for simultaneous spatial transcriptomic and proteomic analysis of a tissue sample. DBiT-seq improves upon previous spatial transcriptomics applications such as High-Definition Spatial Transcriptomics (HDST) and Slide-seq by increasing the number of detectable genes per pixel, increased cellular resolution, and ease of implementation.
VINE-seq is a method to isolate and molecularly characterize the vascular and perivascular cells of the human brain microvessels at single-nuclei resolution. This technique is achieved by combining various known laboratory-based strategies involving the mechanical dissociation of brain tissue samples into single cells, density gradient centrifugation and filtration to isolate nuclei of microvessels, fluorescence-activated cell sorting (FACs) of cellular populations and droplet-based single-nuclei RNA sequencing (drop-snRNA-seq). Altogether, this generates a single-nuclei transcriptomic profile of the various cell types present in the vasculature of the brain. Through processing and analyzing the single-nuclei transcriptomic data, the heterogeneity within and between cell types can be distinguished to construct the molecular landscape of the human brain vasculature that was not previously done before.


