In biochemistry, immunostaining is any use of an antibody-based method to detect a specific protein in a sample. The term "immunostaining" was originally used to refer to the immunohistochemical staining of tissue sections, as first described by Albert Coons in 1941. However, immunostaining now encompasses a broad range of techniques used in histology, cell biology, and molecular biology that use antibody-based staining methods.

Immunofluorescence(IF) is a light microscopy-based technique that allows detection and localization of a wide variety of target biomolecules within a cell or tissue at a quantitative level. The technique utilizes the binding specificity of antibodies and antigens. The specific region an antibody recognizes on an antigen is called an epitope. Several antibodies can recognize the same epitope but differ in their binding affinity. The antibody with the higher affinity for a specific epitope will surpass antibodies with a lower affinity for the same epitope.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.

Hoechst stains are part of a family of blue fluorescent dyes used to stain DNA. These bis-benzimides were originally developed by Hoechst AG, which numbered all their compounds so that the dye Hoechst 33342 is the 33,342nd compound made by the company. There are three related Hoechst stains: Hoechst 33258, Hoechst 33342, and Hoechst 34580. The dyes Hoechst 33258 and Hoechst 33342 are the ones most commonly used and they have similar excitation–emission spectra.

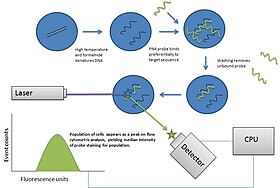
Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only particular parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.
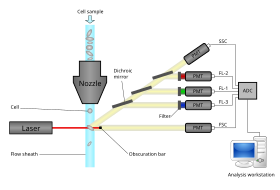
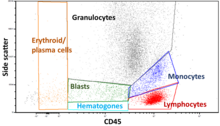
Immunophenotyping is a technique used to study the protein expressed by cells. This technique is commonly used in basic science research and laboratory diagnostic purpose. This can be done on tissue section, cell suspension, etc. An example is the detection of tumor markers, such as in the diagnosis of leukemia. It involves the labelling of white blood cells with antibodies directed against surface proteins on their membrane. By choosing appropriate antibodies, the differentiation of leukemic cells can be accurately determined. The labelled cells are processed in a flow cytometer, a laser-based instrument capable of analyzing thousands of cells per second. The whole procedure can be performed on cells from the blood, bone marrow or spinal fluid in a matter of a few hours.
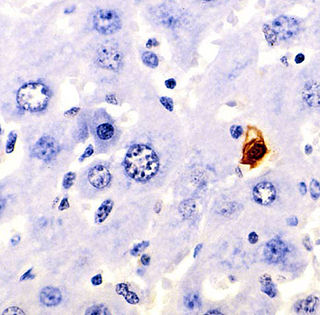
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) is a method for detecting DNA fragmentation by labeling the 3′- hydroxyl termini in the double-strand DNA breaks generated during apoptosis.

Acridine orange is an organic compound that serves as a nucleic acid-selective fluorescent dye with cationic properties useful for cell cycle determination. Acridine orange is cell-permeable, which allows the dye to interact with DNA by intercalation, or RNA via electrostatic attractions. When bound to DNA, acridine orange is very similar spectrally to an organic compound known as fluorescein. Acridine orange and fluorescein have a maximum excitation at 502nm and 525 nm (green). When acridine orange associates with RNA, the fluorescent dye experiences a maximum excitation shift from 525 nm (green) to 460 nm (blue). The shift in maximum excitation also produces a maximum emission of 650 nm (red). Acridine orange is able to withstand low pH environments, allowing the fluorescent dye to penetrate acidic organelles such as lysosomes and phagolysosomes that are membrane-bound organelles essential for acid hydrolysis or for producing products of phagocytosis of apoptotic cells. Acridine orange is used in epifluorescence microscopy and flow cytometry. The ability to penetrate the cell membranes of acidic organelles and cationic properties of acridine orange allows the dye to differentiate between various types of cells. The shift in maximum excitation and emission wavelengths provides a foundation to predict the wavelength at which the cells will stain.

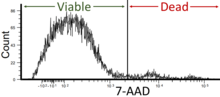
7-Aminoactinomycin D (7-AAD) is a fluorescent chemical compound with a strong affinity for DNA. It is used as a fluorescent marker for DNA in fluorescence microscopy and flow cytometry. It intercalates in double-stranded DNA, with a high affinity for GC-rich regions, making it useful for chromosome banding studies.
Flow-FISH is a cytogenetic technique to quantify the copy number of RNA or specific repetitive elements in genomic DNA of whole cell populations via the combination of flow cytometry with cytogenetic fluorescent in situ hybridization staining protocols.
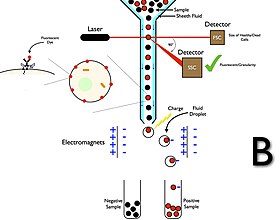
Cell sorting is the process through which a particular cell type is separated from others contained in a sample on the basis of its physical or biological properties, such as size, morphological parameters, viability and both extracellular and intracellular protein expression. The homogeneous cell population obtained after sorting can be used for a variety of applications including research, diagnosis, and therapy.

Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence. Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.
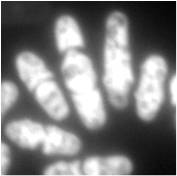
Microfluorimetry is an adaption of fluorimetry for studying the biochemical and biophysical properties of cells by using microscopy to image cell components tagged with fluorescent molecules. It is a type of microphotometry that gives a quantitative measure of the qualitative nature of fluorescent measurement and therefore, allows for definitive results that would have been previously indiscernible to the naked eye.

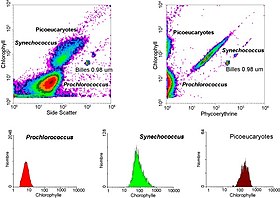
Cytometry is the measurement of number and characteristics of cells. Variables that can be measured by cytometric methods include cell size, cell count, cell morphology, cell cycle phase, DNA content, and the existence or absence of specific proteins on the cell surface or in the cytoplasm. Cytometry is used to characterize and count blood cells in common blood tests such as the complete blood count. In a similar fashion, cytometry is also used in cell biology research and in medical diagnostics to characterize cells in a wide range of applications associated with diseases such as cancer and AIDS.
Cell cycle analysis by DNA content measurement is a method that most frequently employs flow cytometry to distinguish cells in different phases of the cell cycle. Before analysis, the cells are usually permeabilised and treated with a fluorescent dye that stains DNA quantitatively, such as propidium iodide (PI) or 4,6-diamidino-2-phenylindole (DAPI). The fluorescence intensity of the stained cells correlates with the amount of DNA they contain. As the DNA content doubles during the S phase, the DNA content (and thereby intensity of fluorescence) of cells in the G0 phase and G1 phase (before S), in the S phase, and in the G2 phase and M phase (after S) identifies the cell cycle phase position in the major phases (G0/G1 versus S versus G2/M phase) of the cell cycle. The cellular DNA content of individual cells is often plotted as their frequency histogram to provide information about relative frequency (percentage) of cells in the major phases of the cell cycle.
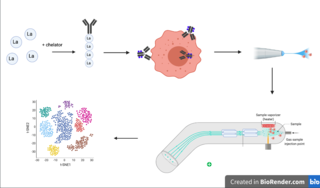
Cytometry by time of flight, or CyTOF, is an application of mass cytometry used to quantify labeled targets on the surface and interior of single cells. CyTOF allows the quantification of multiple cellular components simultaneously using an ICP-MS detector.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
Flow cytometry bioinformatics is the application of bioinformatics to flow cytometry data, which involves storing, retrieving, organizing and analyzing flow cytometry data using extensive computational resources and tools. Flow cytometry bioinformatics requires extensive use of and contributes to the development of techniques from computational statistics and machine learning. Flow cytometry and related methods allow the quantification of multiple independent biomarkers on large numbers of single cells. The rapid growth in the multidimensionality and throughput of flow cytometry data, particularly in the 2000s, has led to the creation of a variety of computational analysis methods, data standards, and public databases for the sharing of results.
Bacterioplankton counting is the estimation of the abundance of bacterioplankton in a specific body of water, which is useful information to marine microbiologists. Various counting methodologies have been developed over the years to determine the number present in the water being observed. Methods used for counting bacterioplankton include epifluorescence microscopy, flow cytometry, measures of productivity through frequency of dividing cells (FDC), thymidine incorporation, and leucine incorporation.
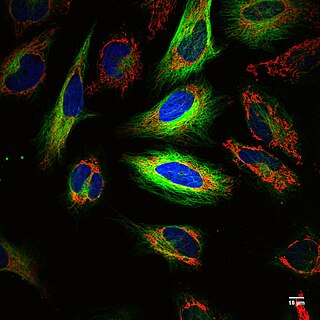
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.