Proteomics is the large-scale study of proteins. Proteins are vital parts of living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.
Glycomics is the comprehensive study of glycomes, including genetic, physiologic, pathologic, and other aspects. Glycomics "is the systematic study of all glycan structures of a given cell type or organism" and is a subset of glycobiology. The term glycomics is derived from the chemical prefix for sweetness or a sugar, "glyco-", and was formed to follow the omics naming convention established by genomics and proteomics.

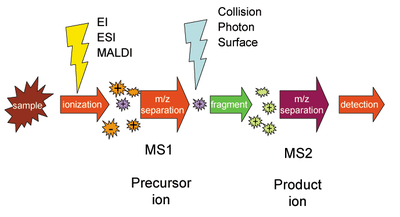
Mass spectrometry (MS) is an analytical technique that is used to measure the mass-to-charge ratio of ions. The results are presented as a mass spectrum, a plot of intensity as a function of the mass-to-charge ratio. Mass spectrometry is used in many different fields and is applied to pure samples as well as complex mixtures.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.

Selected-ion flow-tube mass spectrometry (SIFT-MS) is a quantitative mass spectrometry technique for trace gas analysis which involves the chemical ionization of trace volatile compounds by selected positive precursor ions during a well-defined time period along a flow tube. Absolute concentrations of trace compounds present in air, breath or the headspace of bottled liquid samples can be calculated in real time from the ratio of the precursor and product ion signal ratios, without the need for sample preparation or calibration with standard mixtures. The detection limit of commercially available SIFT-MS instruments extends to the single digit pptv range.

Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
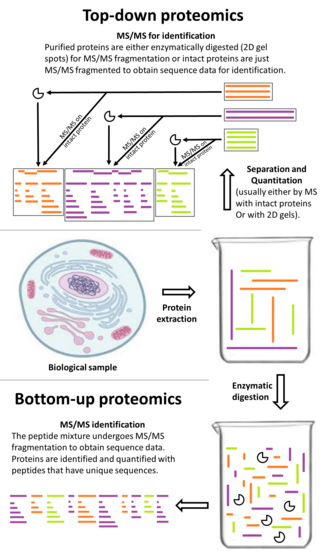
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.
Label-free quantification is a method in mass spectrometry that aims to determine the relative amount of proteins in two or more biological samples. Unlike other methods for protein quantification, label-free quantification does not use a stable isotope containing compound to chemically bind to and thus label the protein.

A triple quadrupole mass spectrometer (TQMS), is a tandem mass spectrometer consisting of two quadrupole mass analyzers in series, with a (non-mass-resolving) radio frequency (RF)–only quadrupole between them to act as a cell for collision-induced dissociation. This configuration is often abbreviated QqQ, here Q1q2Q3.
Unimolecular ion decomposition is the fragmentation of a gas phase ion in a reaction with a molecularity of one. Ions with sufficient internal energy may fragment in a mass spectrometer, which in some cases may degrade the mass spectrometer performance, but in other cases, such as tandem mass spectrometry, the fragmentation can reveal information about the structure of the ion.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with chemical groups that have identical mass (isobaric), but vary in terms of distribution of heavy isotopes in their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy CID (HCD) during tandem mass spectrometry yielding reporter ions of different masses. The most common isobaric tags are amine-reactive tags. However, tags that react with cysteine residues and carbonyl groups have also been described. These amine-reactive groups go through N-hydroxysuccinimide (NHS) reactions, which are based around three types of functional groups. Isobaric labeling methods include tandem mass tags (TMT), isobaric tags for relative and absolute quantification (iTRAQ), mass differential tags for absolute and relative quantification, and dimethyl labeling. TMTs and iTRAQ methods are most common and developed of these methods. Tandem mass tags have a mass reporter region, a cleavable linker region, a mass normalization region, and a protein reactive group and have the same total mass.

Collision-induced dissociation (CID), also known as collisionally activated dissociation (CAD), is a mass spectrometry technique to induce fragmentation of selected ions in the gas phase. The selected ions are usually accelerated by applying an electrical potential to increase the ion kinetic energy and then allowed to collide with neutral molecules. In the collision, some of the kinetic energy is converted into internal energy which results in bond breakage and the fragmentation of the molecular ion into smaller fragments. These fragment ions can then be analyzed by tandem mass spectrometry.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.
In mass spectrometry, data-independent acquisition (DIA) is a method of molecular structure determination in which all ions within a selected m/z range are fragmented and analyzed in a second stage of tandem mass spectrometry. Tandem mass spectra are acquired either by fragmenting all ions that enter the mass spectrometer at a given time or by sequentially isolating and fragmenting ranges of m/z. DIA is an alternative to data-dependent acquisition (DDA) where a fixed number of precursor ions are selected and analyzed by tandem mass spectrometry.
Stable isotope standards and capture by anti-peptide antibodies (SISCAPA) is a mass spectrometry method for measuring the amount of a protein in a biological sample.
Skyline is an open source software for targeted proteomics and metabolomics data analysis. It runs on Microsoft Windows and supports the raw data formats from multiple mass spectrometric vendors. It contains a graphical user interface to display chromatographic data for individual peptide or small molecule analytes.