UPGMA (unweighted pair group method with arithmetic mean) is a simple agglomerative (bottom-up) hierarchical clustering method. It also has a weighted variant, WPGMA, and they are generally attributed to Sokal and Michener.[1]
Note that the unweighted term indicates that all distances contribute equally to each average that is computed and does not refer to the math by which it is achieved. Thus the simple averaging in WPGMA produces a weighted result and the proportional averaging in UPGMA produces an unweighted result (see the working example).[2]
Algorithm
The UPGMA algorithm constructs a rooted tree (dendrogram) that reflects the structure present in a pairwise similarity matrix (or a dissimilarity matrix). At each step, the nearest two clusters are combined into a higher-level cluster. The distance between any two clusters and , each of size (i.e., cardinality) and , is taken to be the average of all distances between pairs of objects in and in , that is, the mean distance between elements of each cluster:
In other words, at each clustering step, the updated distance between the joined clusters and a new cluster is given by the proportional averaging of the and distances:
The UPGMA algorithm produces rooted dendrograms and requires a constant-rate assumption - that is, it assumes an ultrametric tree in which the distances from the root to every branch tip are equal. When the tips are molecular data (i.e., DNA, RNA and protein) sampled at the same time, the ultrametricity assumption becomes equivalent to assuming a molecular clock.
Let us assume that we have five elements and the following matrix of pairwise distances between them:
a
b
c
d
e
a
0
17
21
31
23
b
17
0
30
34
21
c
21
30
0
28
39
d
31
34
28
0
43
e
23
21
39
43
0
In this example, is the smallest value of , so we join elements and .
First branch length estimation
Let denote the node to which and are now connected. Setting ensures that elements and are equidistant from . This corresponds to the expectation of the ultrametricity hypothesis. The branches joining and to then have lengths (see the final dendrogram)
First distance matrix update
We then proceed to update the initial distance matrix into a new distance matrix (see below), reduced in size by one row and one column because of the clustering of with . Bold values in correspond to the new distances, calculated by averaging distances between each element of the first cluster and each of the remaining elements:
Italicized values in are not affected by the matrix update as they correspond to distances between elements not involved in the first cluster.
Second step
Second clustering
We now reiterate the three previous steps, starting from the new distance matrix
(a,b)
c
d
e
(a,b)
0
25.5
32.5
22
c
25.5
0
28
39
d
32.5
28
0
43
e
22
39
43
0
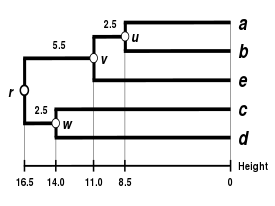
Here, is the smallest value of , so we join cluster and element .
Second branch length estimation
Let denote the node to which and are now connected. Because of the ultrametricity constraint, the branches joining or to , and to are equal and have the following length:
We then proceed to update into a new distance matrix (see below), reduced in size by one row and one column because of the clustering of with . Bold values in correspond to the new distances, calculated by proportional averaging:
Thanks to this proportional average, the calculation of this new distance accounts for the larger size of the cluster (two elements) with respect to (one element). Similarly:
Proportional averaging therefore gives equal weight to the initial distances of matrix . This is the reason why the method is unweighted, not with respect to the mathematical procedure but with respect to the initial distances.
Third step
Third clustering
We again reiterate the three previous steps, starting from the updated distance matrix .
((a,b),e)
c
d
((a,b),e)
0
30
36
c
30
0
28
d
36
28
0
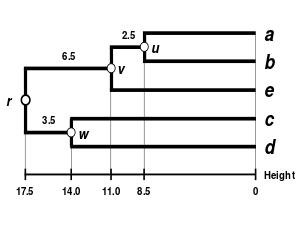
Here, is the smallest value of , so we join elements and .
Third branch length estimation
Let denote the node to which and are now connected. The branches joining and to then have lengths (see the final dendrogram)
Third distance matrix update
There is a single entry to update, keeping in mind that the two elements and each have a contribution of in the average computation:
Final step
The final matrix is:
((a,b),e)
(c,d)
((a,b),e)
0
33
(c,d)
33
0
So we join clusters and .
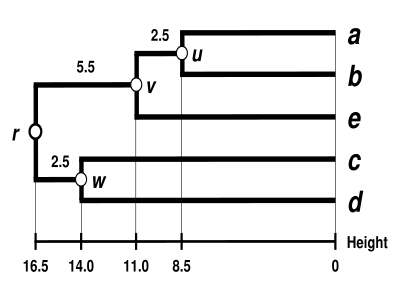
Let denote the (root) node to which and are now connected. The branches joining and to then have lengths:
We deduce the two remaining branch lengths:
The UPGMA dendrogram
The dendrogram is now complete.[5] It is ultrametric because all tips ( to ) are equidistant from :
The dendrogram is therefore rooted by , its deepest node.
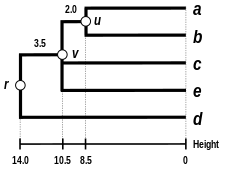
Comparison with other linkages
Alternative linkage schemes include single linkage clustering, complete linkage clustering, and WPGMA average linkage clustering. Implementing a different linkage is simply a matter of using a different formula to calculate inter-cluster distances during the distance matrix update steps of the above algorithm. Complete linkage clustering avoids a drawback of the alternative single linkage clustering method - the so-called chaining phenomenon, where clusters formed via single linkage clustering may be forced together due to single elements being close to each other, even though many of the elements in each cluster may be very distant to each other. Complete linkage tends to find compact clusters of approximately equal diameters.[6]
Comparison of dendrograms obtained under different clustering methods from the same distance matrix.
In ecology, it is one of the most popular methods for the classification of sampling units (such as vegetation plots) on the basis of their pairwise similarities in relevant descriptor variables (such as species composition).[7] For example, it has been used to understand the trophic interaction between marine bacteria and protists.[8]
In bioinformatics, UPGMA is used for the creation of phenetictrees (phenograms). UPGMA was initially designed for use in protein electrophoresis studies, but is currently most often used to produce guide trees for more sophisticated algorithms. This algorithm is for example used in sequence alignment procedures, as it proposes one order in which the sequences will be aligned. Indeed, the guide tree aims at grouping the most similar sequences, regardless of their evolutionary rate or phylogenetic affinities, and that is exactly the goal of UPGMA[9]
In phylogenetics, UPGMA assumes a constant rate of evolution (molecular clock hypothesis) and that all sequences were sampled at the same time, and is not a well-regarded method for inferring relationships unless this assumption has been tested and justified for the data set being used. Notice that even under a 'strict clock', sequences sampled at different times should not lead to an ultrametric tree.
Time complexity
A trivial implementation of the algorithm to construct the UPGMA tree has time complexity, and using a heap for each cluster to keep its distances from other cluster reduces its time to . Fionn Murtagh presented an time and space algorithm.[10]
The moment of inertia, otherwise known as the mass moment of inertia, angular mass, second moment of mass, or most accurately, rotational inertia, of a rigid body is a quantity that determines the torque needed for a desired angular acceleration about a rotational axis, akin to how mass determines the force needed for a desired acceleration. It depends on the body's mass distribution and the axis chosen, with larger moments requiring more torque to change the body's rate of rotation.
In mathematics, an ultrametric space is a metric space in which the triangle inequality is strengthened to . Sometimes the associated metric is also called a non-Archimedean metric or super-metric.
In bioinformatics, neighbor joining is a bottom-up (agglomerative) clustering method for the creation of phylogenetic trees, created by Naruya Saitou and Masatoshi Nei in 1987. Usually based on DNA or protein sequence data, the algorithm requires knowledge of the distance between each pair of taxa to create the phylogenetic tree.
In data mining and statistics, hierarchical clustering is a method of cluster analysis that seeks to build a hierarchy of clusters. Strategies for hierarchical clustering generally fall into two categories:
Cluster analysis or clustering is the task of grouping a set of objects in such a way that objects in the same group are more similar to each other than to those in other groups (clusters). It is a main task of exploratory data analysis, and a common technique for statistical data analysis, used in many fields, including pattern recognition, image analysis, information retrieval, bioinformatics, data compression, computer graphics and machine learning.
Verlet integration is a numerical method used to integrate Newton's equations of motion. It is frequently used to calculate trajectories of particles in molecular dynamics simulations and computer graphics. The algorithm was first used in 1791 by Jean Baptiste Delambre and has been rediscovered many times since then, most recently by Loup Verlet in the 1960s for use in molecular dynamics. It was also used by P. H. Cowell and A. C. C. Crommelin in 1909 to compute the orbit of Halley's Comet, and by Carl Størmer in 1907 to study the trajectories of electrical particles in a magnetic field . The Verlet integrator provides good numerical stability, as well as other properties that are important in physical systems such as time reversibility and preservation of the symplectic form on phase space, at no significant additional computational cost over the simple Euler method.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.
Multi-task learning (MTL) is a subfield of machine learning in which multiple learning tasks are solved at the same time, while exploiting commonalities and differences across tasks. This can result in improved learning efficiency and prediction accuracy for the task-specific models, when compared to training the models separately. Early versions of MTL were called "hints".
In applied statistics, total least squares is a type of errors-in-variables regression, a least squares data modeling technique in which observational errors on both dependent and independent variables are taken into account. It is a generalization of Deming regression and also of orthogonal regression, and can be applied to both linear and non-linear models.
The Lenstra–Lenstra–Lovász (LLL) lattice basis reduction algorithm is a polynomial time lattice reduction algorithm invented by Arjen Lenstra, Hendrik Lenstra and László Lovász in 1982. Given a basis with n-dimensional integer coordinates, for a lattice L with , the LLL algorithm calculates an LLL-reduced lattice basis in time
In mathematics, differential algebra is, broadly speaking, the area of mathematics consisting in the study of differential equations and differential operators as algebraic objects in view of deriving properties of differential equations and operators without computing the solutions, similarly as polynomial algebras are used for the study of algebraic varieties, which are solution sets of systems of polynomial equations. Weyl algebras and Lie algebras may be considered as belonging to differential algebra.
Quantum walks are quantum analogues of classical random walks. In contrast to the classical random walk, where the walker occupies definite states and the randomness arises due to stochastic transitions between states, in quantum walks randomness arises through: (1) quantum superposition of states, (2) non-random, reversible unitary evolution and (3) collapse of the wave function due to state measurements.
In statistics, single-linkage clustering is one of several methods of hierarchical clustering. It is based on grouping clusters in bottom-up fashion, at each step combining two clusters that contain the closest pair of elements not yet belonging to the same cluster as each other.
In mathematics, the Johnson–Lindenstrauss lemma is a result named after William B. Johnson and Joram Lindenstrauss concerning low-distortion embeddings of points from high-dimensional into low-dimensional Euclidean space. The lemma states that a set of points in a high-dimensional space can be embedded into a space of much lower dimension in such a way that distances between the points are nearly preserved. The map used for the embedding is at least Lipschitz, and can even be taken to be an orthogonal projection.
Complete-linkage clustering is one of several methods of agglomerative hierarchical clustering. At the beginning of the process, each element is in a cluster of its own. The clusters are then sequentially combined into larger clusters until all elements end up being in the same cluster. The method is also known as farthest neighbour clustering. The result of the clustering can be visualized as a dendrogram, which shows the sequence of cluster fusion and the distance at which each fusion took place.
In the theory of cluster analysis, the nearest-neighbor chain algorithm is an algorithm that can speed up several methods for agglomerative hierarchical clustering. These are methods that take a collection of points as input, and create a hierarchy of clusters of points by repeatedly merging pairs of smaller clusters to form larger clusters. The clustering methods that the nearest-neighbor chain algorithm can be used for include Ward's method, complete-linkage clustering, and single-linkage clustering; these all work by repeatedly merging the closest two clusters but use different definitions of the distance between clusters. The cluster distances for which the nearest-neighbor chain algorithm works are called reducible and are characterized by a simple inequality among certain cluster distances.
In computational learning theory, Occam learning is a model of algorithmic learning where the objective of the learner is to output a succinct representation of received training data. This is closely related to probably approximately correct (PAC) learning, where the learner is evaluated on its predictive power of a test set.
WPGMA is a simple agglomerative (bottom-up) hierarchical clustering method, generally attributed to Sokal and Michener.
A central problem in algorithmic graph theory is the shortest path problem. One of the generalizations of the shortest path problem is known as the single-source-shortest-paths (SSSP) problem, which consists of finding the shortest paths from a source vertex to all other vertices in the graph. There are classical sequential algorithms which solve this problem, such as Dijkstra's algorithm. In this article, however, we present two parallel algorithms solving this problem.
In the context of quantum computing, the quantum walk search is a quantum algorithm for finding a marked node in a graph.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.