Carl Richard Woese was an American microbiologist and biophysicist. Woese is famous for defining the Archaea in 1977 through a pioneering phylogenetic taxonomy of 16S ribosomal RNA, a technique that has revolutionized microbiology. He also originated the RNA world hypothesis in 1967, although not by that name. Woese held the Stanley O. Ikenberry Chair and was professor of microbiology at the University of Illinois Urbana–Champaign.
In biological taxonomy, a domain, also dominion, superkingdom, realm, or empire, is the highest taxonomic rank of all organisms taken together. It was introduced in the three-domain system of taxonomy devised by Carl Woese, Otto Kandler and Mark Wheelis in 1990.

Nanoarchaeota is a proposed phylum in the domain Archaea that currently has only one representative, Nanoarchaeum equitans, which was discovered in a submarine hydrothermal vent and first described in 2002.

The Thermoproteota are prokaryotes that have been classified as a phylum of the Archaea domain. Initially, the Thermoproteota were thought to be sulfur-dependent extremophiles but recent studies have identified characteristic Thermoproteota environmental rRNA indicating the organisms may be the most abundant archaea in the marine environment. Originally, they were separated from the other archaea based on rRNA sequences; other physiological features, such as lack of histones, have supported this division, although some crenarchaea were found to have histones. Until recently all cultured Thermoproteota had been thermophilic or hyperthermophilic organisms, some of which have the ability to grow at up to 113°C. These organisms stain Gram negative and are morphologically diverse, having rod, cocci, filamentous and oddly-shaped cells.

The Korarchaeota is a proposed phylum within the Archaea. The name is derived from the Greek noun koros or kore, meaning young man or young woman, and the Greek adjective archaios which means ancient. They are also known as Xenarchaeota. The name is equivalent to Candidatus Korarchaeota, and they go by the name Xenarchaeota or Xenarchaea as well.

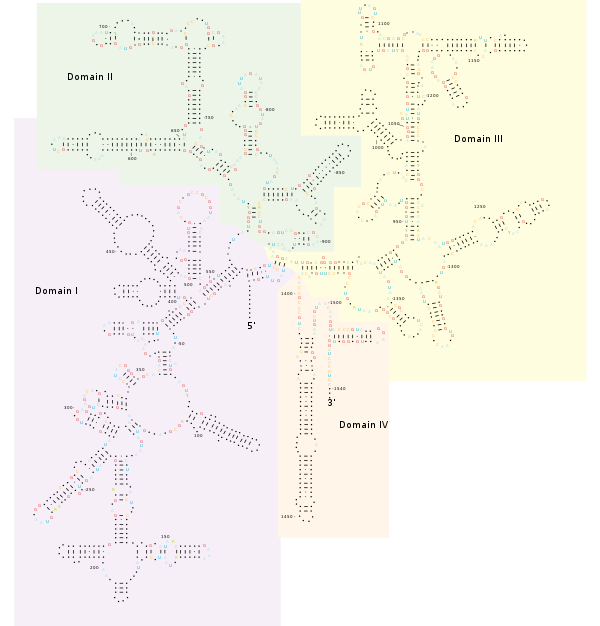
Ribosomal DNA (rDNA) is a DNA sequence that codes for ribosomal RNA. These sequences regulate transcription initiation and amplification, and contain both transcribed and non-transcribed spacer segments.
Internal transcribed spacer (ITS) is the spacer DNA situated between the small-subunit ribosomal RNA (rRNA) and large-subunit rRNA genes in the chromosome or the corresponding transcribed region in the polycistronic rRNA precursor transcript.

Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.

George Edward Fox is an astrobiologist, a Professor Emeritus and researcher at the University of Houston. He is an elected fellow of the American Academy of Microbiology, the American Association for the Advancement of Science, American Institute for Medical and Biological Engineering and the International Astrobiology Society. Fox received his B.A. degree in 1967, and completed his Ph.D. degree in 1974; both in chemical engineering at Syracuse University.

Archaea is a domain of single-celled organisms. These microorganisms lack cell nuclei and are therefore prokaryotes. Archaea were initially classified as bacteria, receiving the name archaebacteria, but this term has fallen out of use.
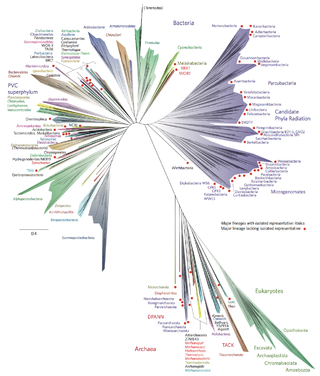
Bacterial phyla constitute the major lineages of the domain Bacteria. While the exact definition of a bacterial phylum is debated, a popular definition is that a bacterial phylum is a monophyletic lineage of bacteria whose 16S rRNA genes share a pairwise sequence identity of ~75% or less with those of the members of other bacterial phyla.
Bacterial taxonomy is subfield of taxonomy devoted to the classification of bacteria specimens into taxonomic ranks.
There are several models of the Branching order of bacterial phyla, one of these was proposed in 1987 paper by Carl Woese.

'The All-Species Living Tree' Project is a collaboration between various academic groups/institutes, such as ARB, SILVA rRNA database project, and LPSN, with the aim of assembling a database of 16S rRNA sequences of all validly published species of Bacteria and Archaea. At one stage, 23S sequences were also collected, but this has since stopped.
Conserved signature inserts and deletions (CSIs) in protein sequences provide an important category of molecular markers for understanding phylogenetic relationships. CSIs, brought about by rare genetic changes, provide useful phylogenetic markers that are generally of defined size and they are flanked on both sides by conserved regions to ensure their reliability. While indels can be arbitrary inserts or deletions, CSIs are defined as only those protein indels that are present within conserved regions of the protein.
Community fingerprinting is a set of molecular biology techniques that can be used to quickly profile the diversity of a microbial community. Rather than directly identifying or counting individual cells in an environmental sample, these techniques show how many variants of a gene are present. In general, it is assumed that each different gene variant represents a different type of microbe. Community fingerprinting is used by microbiologists studying a variety of microbial systems to measure biodiversity or track changes in community structure over time. The method analyzes environmental samples by assaying genomic DNA. This approach offers an alternative to microbial culturing, which is important because most microbes cannot be cultured in the laboratory. Community fingerprinting does not result in identification of individual microbe species; instead, it presents an overall picture of a microbial community. These methods are now largely being replaced by high throughput sequencing, such as targeted microbiome analysis and metagenomics.

The eocyte hypothesis in evolutionary biology proposes that the eukaryotes originated from a group of prokaryotes called eocytes. After his team at the University of California, Los Angeles discovered eocytes in 1984, James A. Lake formulated the hypothesis as "eocyte tree" that proposed eukaryotes as part of archaea. Lake hypothesised the tree of life as having only two primary branches: prokaryotes, which include Bacteria and Archaea, and karyotes, that comprise Eukaryotes and eocytes. Parts of this early hypothesis were revived in a newer two-domain system of biological classification which named the primary domains as Archaea and Bacteria.
Microbial phylogenetics is the study of the manner in which various groups of microorganisms are genetically related. This helps to trace their evolution. To study these relationships biologists rely on comparative genomics, as physiology and comparative anatomy are not possible methods.
Microbial DNA barcoding is the use of DNA metabarcoding to characterize a mixture of microorganisms. DNA metabarcoding is a method of DNA barcoding that uses universal genetic markers to identify DNA of a mixture of organisms.
Thermodesulfobacterium hveragerdense is a bacterial species belonging to genus Thermodesulfobacterium, which are thermophilic sulfate-reducing bacteria. This species is found in aquatic areas of high temperature, and lives in freshwater like most, but not all Thermodesulfobacterium species It was first isolated from hotsprings in Iceland.