Related Research Articles
Prader–Willi syndrome (PWS) is a genetic disorder caused by a loss of function of specific genes on chromosome 15. In newborns, symptoms include weak muscles, poor feeding, and slow development. Beginning in childhood, those affected become constantly hungry, which often leads to obesity and type 2 diabetes. Mild to moderate intellectual impairment and behavioral problems are also typical of the disorder. Often, affected individuals have a narrow forehead, small hands and feet, short height, and light skin and hair. Most are unable to have children.
Autism spectrum disorders (ASD) are neurodevelopmental disorders that begin in early childhood, persist throughout adulthood, and affect three crucial areas of development: communication, social interaction and restricted patterns of behavior. There are many conditions comorbid to autism spectrum disorders such as attention-deficit hyperactivity disorder and epilepsy.
Smith–Magenis Syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.

The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis. Although the genetics of autism are complex, autism spectrum disorder (ASD) is explained more by multigene effects than by rare mutations with large effects.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

2q37 deletion syndrome is a disorder caused by the deletion of a small piece of chromosome 2 in which one or more of 3 sub-bands, 2q37.1, 2q37.2, and 2q37.3, of the last band of one of the chromosome 2’s long arms are deleted. The first report of this disorder was in 1989.

Angelman syndrome or Angelman's syndrome (AS) is a genetic disorder that mainly affects the nervous system. Symptoms include a small head and a specific facial appearance, severe intellectual disability, developmental disability, limited to no functional speech, balance and movement problems, seizures, and sleep problems. Children usually have a happy personality and have a particular interest in water. The symptoms generally become noticeable by one year of age.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Potocki–Lupski syndrome (PTLS), also known as dup(17)p11.2p11.2 syndrome, trisomy 17p11.2 or duplication 17p11.2 syndrome, is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). The duplication was first described as a case study in 1996. In 2000, the first study of the disease was released, and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. PTLS is named for two researchers involved in the latter phases, Drs. Lorraine Potocki and James R. Lupski of Baylor College of Medicine.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt-Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.
1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.
1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.

The imprinted brain hypothesis is an unsubstantiated hypothesis in evolutionary psychology regarding the causes of autism spectrum and schizophrenia spectrum disorders, first presented by Bernard Crespi and Christopher Badcock in 2008. It claims that certain autistic and schizotypal traits are opposites, and that this implies the etiology of the two conditions must be at odds.
Burnside–Butler syndrome is a name that has been applied to the effects of microdeletion of DNA sequences involving four neurodevelopmental genes. Varying developmental and psychiatric disorders have been attributed to the microdeletion; however, the great majority of people with the deletion do not have any clinical features associated with it. More studies are needed to delineate the range of clinical presentation.
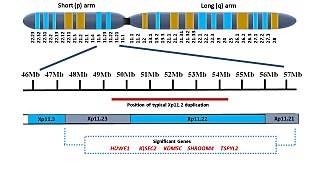
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.

DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, intellectual disability and cleft palate. Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.
Beck–Fahrner syndrome, also known as BEFAHRS and TET3 deficiency, is a rare genetic disorder caused by mutations of the TET3 gene. It can occur de novo or can be inherited in an autosomal dominant manner. Mutations in the TET3 gene disrupts DNA demethylation during early embryogenesis and neural development. Most common clinical presentation includes global developmental delay, psychomotor retardation, neurodevelopmental disorders, hypotonia, epilepsy and dysmorphic features. It is diagnosed using molecular and genetic testing in setting of typical symptoms. Management is supportive and intended to improve quality of life.
References
- 1 2 3 4 5 6 7 8 9 10 11 Taylor, Cora M.; Smith, Rebecca; et al. (1993). "16p11.2 Recurrent Deletion". GeneReviews. University of Washington, Seattle. PMID 20301775.
- 1 2 3 4 Chung, Wendy K; Roberts, Timothy PL; et al. (1 June 2021). "16p11.2 deletion syndrome". Current Opinion in Genetics & Development. 68: 49–56. doi:10.1016/j.gde.2021.01.011. ISSN 0959-437X. PMC 10256135 . PMID 33667823.
- 1 2 Weiss, Lauren A.; Shen, Yiping; et al. (14 February 2008). "Association between Microdeletion and Microduplication at 16p11.2 and Autism". New England Journal of Medicine. 358 (7): 667–675. doi: 10.1056/NEJMoa075974 . PMID 18184952.
- 1 2 3 Fetit, Rana; Price, David J.; et al. (October 2020). "Understanding the clinical manifestations of 16p11.2 deletion syndrome: a series of developmental case reports in children". Psychiatric Genetics. 30 (5): 136–140. doi:10.1097/YPG.0000000000000259. ISSN 0955-8829. PMC 7497286 . PMID 32732550.
- 1 2 3 Steinman, Kyle J.; Spence, Sarah J.; et al. (November 2016). "16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort". American Journal of Medical Genetics Part A. 170 (11): 2943–2955. doi:10.1002/ajmg.a.37820. PMID 27410714. S2CID 2469192.
- ↑ Zufferey, Flore; Sherr, Elliott H; et al. (October 2012). "A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders". Journal of Medical Genetics. 49 (10): 660–668. doi: 10.1136/jmedgenet-2012-101203 . PMC 3494011 . PMID 23054248.
- ↑ Gill, Richard; Chen, Qixuan; et al. (December 2014). "Eating in the absence of hunger but not loss of control behaviors are associated with 16p11.2 deletions: 16p11.2 Deletion and Disinhibited Eating". Obesity. 22 (12): 2625–2631. doi: 10.1002/oby.20892 . PMID 25234362.
- ↑ Egolf, Laura E.; Vaksman, Zalman; et al. (September 2019). "Germline 16p11.2 Microdeletion Predisposes to Neuroblastoma". The American Journal of Human Genetics. 105 (3): 658–668. doi: 10.1016/j.ajhg.2019.07.020 . PMC 6731370 . PMID 31474320.
- ↑ Chung, Wendy K. (12 October 2011). "An Overview of Monogenic and Syndromic Obesities in Humans". Pediatr Blood Cancer. 58 (1): 122–128. doi:10.1002/pbc.23372. ISSN 1545-5009. PMC 3215910 . PMID 21994130.
- ↑ Kleinendorst, Lotte; van den Heuvel, Lieke M.; et al. (September 2020). "Who ever heard of 16p11.2 deletion syndrome? Parents' perspectives on a susceptibility copy number variation syndrome". European Journal of Human Genetics. 28 (9): 1196–1204. doi:10.1038/s41431-020-0644-6. ISSN 1476-5438. PMC 7608422 . PMID 32415274.
- ↑ Szelest, Monika; Stefaniak, Martyna; et al. (10 March 2021). "Three case reports of patients indicating the diversity of molecular and clinical features of 16p11.2 microdeletion anomaly". BMC Medical Genomics. 14 (1): 76. doi: 10.1186/s12920-021-00929-8 . ISSN 1755-8794. PMC 7945342 . PMID 33691695.