Related Research Articles
Angela Vincent is Emeritus professor at the University of Oxford and a Fellow of Somerville College, Oxford.

Lambert–Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder characterized by muscle weakness of the limbs.
Neuromyotonia (NMT) is a form of peripheral nerve hyperexcitability that causes spontaneous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. NMT along with Morvan's syndrome are the most severe types in the Peripheral Nerve Hyperexciteability spectrum. Example of two more common and less severe syndromes in the spectrum are Cramp Fasciculation Syndrome and Benign Fasciculation Syndrome. NMT can have both hereditary and acquired forms. The prevalence of NMT is unknown.
Morvan's syndrome is a rare, life-threatening autoimmune disease named after the nineteenth century French physician Augustin Marie Morvan. "La chorée fibrillaire" was first coined by Morvan in 1890 when describing patients with multiple, irregular contractions of the long muscles, cramping, weakness, pruritus, hyperhidrosis, insomnia and delirium. It normally presents with a slow insidious onset over months to years. Approximately 90% of cases spontaneously go into remission, while the other 10% of cases lead to death.
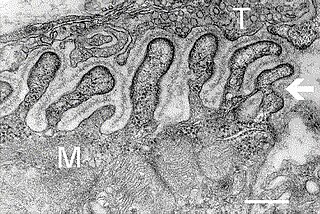
A neuromuscular junction is a chemical synapse between a motor neuron and a muscle fiber.

Stiff-person syndrome (SPS), also known as stiff-man syndrome, is a rare neurologic disorder of unclear cause characterized by progressive muscular rigidity and stiffness. The stiffness primarily affects the truncal muscles and is superimposed by spasms, resulting in postural deformities. Chronic pain, impaired mobility, and lumbar hyperlordosis are common symptoms.
Paraneoplastic cerebellar degeneration (PCD) is a paraneoplastic syndrome associated with a broad variety of tumors including lung cancer, ovarian cancer, breast cancer, Hodgkin’s lymphoma and others. PCD is a rare condition that occurs in less than 1% of cancer patients.
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where affected individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood. GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC). There are at least six types of GEFS+, delineated by their causative gene. Known causative gene mutations are in the sodium channel α subunit genes SCN1A, an associated β subunit SCN1B, and in a GABAA receptor γ subunit gene, in GABRG2 and there is another gene related with calcium channel the PCDH19 which is also known as Epilepsy Female with Mental Retardation. Penetrance for this disorder is estimated at 60%.
Hashimoto's encephalopathy, also known as steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT), is a neurological condition characterized by encephalopathy, thyroid autoimmunity, and good clinical response to corticosteroids. It is associated with Hashimoto's thyroiditis, and was first described in 1966. It is sometimes referred to as a neuroendocrine disorder, although the condition's relationship to the endocrine system is widely disputed. It is recognized as a rare disease by the NIH Genetic and Rare Diseases Information Center.

Limbic encephalitis is a form of encephalitis, a disease characterized by inflammation of the brain. Limbic encephalitis is caused by autoimmunity: an abnormal state where the body produces antibodies against itself. Some cases are associated with cancer and some are not. Although the disease is known as "limbic" encephalitis, it is seldom limited to the limbic system and post-mortem studies usually show involvement of other parts of the brain. The disease was first described by Brierley and others in 1960 as a series of three cases. The link to cancer was first noted in 1968 and confirmed by later investigators.
A paraneoplastic syndrome is a syndrome that is the consequence of a tumor in the body. It is specifically due to the production of chemical signaling molecules by tumor cells or by an immune response against the tumor. Unlike a mass effect, it is not due to the local presence of cancer cells.
Anti-glutamate receptor antibodies are autoantibodies detected in serum and/or cerebrospinal fluid samples of a variety of disorders such as encephalitis, epilepsy and ataxia. Clinical and experimental studies starting around the year 2000 suggest that these antibodies are not simply epiphenomena and are involved in autoimmune disease pathogenesis.

Leucine-rich, glioma inactivated 1, also known as LGI1, is a protein which in humans is encoded by the LGI1 gene. It may be a metastasis suppressor.
Neuromuscular junction disease is a medical condition where the normal conduction through the neuromuscular junction fails to function correctly.
Ian Kirkland Hart FRCP was a lecturer and consultant in neurology at the Walton Centre in Liverpool. He ran a clinic for neurological paraneoplastic syndromes, myasthenia gravis, neuromyotonia, Lambert–Eaton myasthenic syndrome and autoimmune encephalitis. He was also the founder member of the Walton Centre Clinical Neuroimmunology Group researching on autoantibody-associated neurological diseases.

Anti-NMDA receptor encephalitis is a type of brain inflammation caused by antibodies. Early symptoms may include fever, headache, and feeling tired. This is then typically followed by psychosis which presents with false beliefs (delusions) and seeing or hearing things that others do not see or hear (hallucinations). People are also often agitated or confused. Over time, seizures, decreased breathing, and blood pressure and heart rate variability typically occur. In some cases, patients may develop catatonia.
Bickerstaff brainstem encephalitis is a rare inflammatory disorder of the central nervous system, first described by Edwin Bickerstaff in 1951. It may also affect the peripheral nervous system, and has features in common with both Miller Fisher syndrome and Guillain–Barré syndrome.

Autoimmune encephalitis (AIE) is a type of encephalitis, and one of the most common causes of noninfectious encephalitis. It can be triggered by tumors, infections, or it may be cryptogenic. The neurological manifestations can be either acute or subacute and usually develop within six weeks. The clinical manifestations include behavioral and psychiatric symptoms, autonomic disturbances, movement disorders, and seizures.

Autoimmune autonomic ganglionopathy is a type of immune-mediated autonomic failure that is associated with antibodies against the ganglionic nicotinic acetylcholine receptor present in sympathetic, parasympathetic, and enteric ganglia. Typical symptoms include gastrointestinal dysmotility, orthostatic hypotension, and tonic pupils. Many cases have a sudden onset, but others worsen over time, resembling degenerative forms of autonomic dysfunction. For milder cases, supportive treatment is used to manage symptoms. Plasma exchange, intravenous immunoglobulin, corticosteroids, or immunosuppression have been used successfully to treat more severe cases.
Anti-Hu associated encephalitis, also known as Anti-ANNA1 associated encephalitis, is an uncommon form of brain inflammation that is associated with an underlying cancer. It can cause psychiatric symptoms such as depression, anxiety, and hallucinations. It can also produce neurological symptoms such as confusion, memory loss, weakness, sensory loss, pain, seizures, and problems coordinating the movement of the body.
References
- ↑ Rojas, Galeno; Demey, Ignacio; Quiroga, Julieta; Cejas, Luciana Leon; Bonardo, Pablo; Roca, Claudia Uribe; Parisi, Virginia Laura; Gatto, Emilia; Rugilo, Carlos; Ollari, Juan; Pardal, Manuel Fernandez (2016-04-05). "VGKC-Complex Antibody Encephalitis: Clinical Manifestations and Response to Immunotherapy (P2.251)". Neurology. 86 (16 Supplement). ISSN 0028-3878.
- 1 2 Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, et al. (1997). "Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia". Ann Neurol. 41 (2): 238–46. doi:10.1002/ana.410410215. PMID 9029073. S2CID 30375137.
- ↑ Vincent A (2008). "Autoimmune channelopathies: John Newsom-Davis's work and legacy. A summary of the Newsom-Davis Memorial Lecture 2008". J Neuroimmunol. 201–202: 245–9. doi:10.1016/j.jneuroim.2008.07.007. PMID 18722023. S2CID 28767520.
- 1 2 3 4 5 Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. (2010). "Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia". Brain. 133 (9): 2734–48. doi:10.1093/brain/awq213. PMC 2929337 . PMID 20663977.
- ↑ Klein CJ, Lennon VA, Aston PA, McKeon A, O'Toole O, Quek A, et al. (2013). "Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping". JAMA Neurol. 70 (2): 229–34. doi:10.1001/jamaneurol.2013.592. PMC 3895328 . PMID 23407760.
- 1 2 3 4 Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. (2010). "Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series". Lancet Neurol. 9 (8): 776–85. doi:10.1016/S1474-4422(10)70137-X. PMC 3086669 . PMID 20580615.
- ↑ Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. (2011). "Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis". Ann Neurol. 69 (5): 892–900. doi:10.1002/ana.22307. PMID 21416487. S2CID 13775077.
- 1 2 3 4 Dalmau J, Rosenfeld MR (2014). "Autoimmune encephalitis update". Neuro Oncol. 16 (6): 771–8. doi:10.1093/neuonc/nou030. PMC 4022229 . PMID 24637228.
- ↑ Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, et al. (2010). "Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy". Proc Natl Acad Sci U S A. 107 (8): 3799–804. Bibcode:2010PNAS..107.3799F. doi: 10.1073/pnas.0914537107 . PMC 2840530 . PMID 20133599.
- ↑ Gregor A, Albrecht B, Bader I, Bijlsma EK, Ekici AB, Engels H, et al. (2011). "Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1". BMC Med Genet. 12: 106. doi: 10.1186/1471-2350-12-106 . PMC 3162517 . PMID 21827697.
- ↑ Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. (2004). "Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis". Brain. 127 (Pt 3): 701–12. doi: 10.1093/brain/awh077 . PMID 14960497.
- ↑ Dalmau, J Rosenfeld, MR. Paraneoplastic and autoimmune encephalitis. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on October 11, 2015.)
- ↑ Somnier, F. E. (Apr 2015). Autoimmune encephalitis History & current knowledge (PDF). Copenhagen, Denmark: Statens Serum Institut. p. 6. Retrieved 2015-10-12.