
Megalencephaly is a growth development disorder in which the brain is abnormally large. It is characterized by a brain with an average weight that is 2.5 standard deviations above the mean of the general population. Approximately 1 out of 50 children (2%) are said to have the characteristics of megalencephaly in the general population.
Autism spectrum disorders (ASD) are neurodevelopmental disorders that begin in early childhood, persist throughout adulthood, and affect three crucial areas of development: communication, social interaction and restricted patterns of behavior. There are many conditions comorbid to autism spectrum disorders such as attention-deficit hyperactivity disorder and epilepsy.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness. It was described by John Menkes in the 1950s.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.
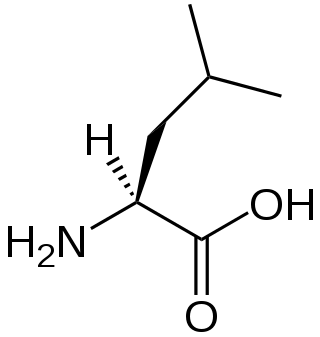
A branched-chain amino acid (BCAA) is an amino acid having an aliphatic side-chain with a branch. Among the proteinogenic amino acids, there are three BCAAs: leucine, isoleucine, and valine. Non-proteinogenic BCAAs include 2-aminoisobutyric acid and alloisoleucine.
Neurodevelopmental disorders are a group of neurological disorders that affect the development of the nervous system, leading to abnormal brain function which may affect emotion, learning ability, self-control, and memory. The effects of neurodevelopmental disorders tend to last for a person's lifetime.
The branched-chain α-ketoacid dehydrogenase complex is a multi-subunit complex of enzymes that is found on the mitochondrial inner membrane. This enzyme complex catalyzes the oxidative decarboxylation of branched, short-chain alpha-ketoacids. BCKDC is a member of the mitochondrial α-ketoacid dehydrogenase complex family comprising pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, key enzymes that function in the Krebs cycle.
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.
2-Methylbutyryl-CoA dehydrogenase deficiency is an autosomal recessive metabolic disorder. It causes the body to be unable to process the amino acid isoleucine properly. Initial case reports identified individuals with developmental delay and epilepsy, however most cases identified through newborn screening have been asymptomatic.

Succinic semialdehyde dehydrogenase deficiency (SSADHD) is a rare autosomal recessive disorder of the degradation pathway of the inhibitory neurotransmitter γ-aminobutyric acid, or GABA. The disorder has been identified in approximately 350 families, with a significant proportion being consanguineous families. The first case was identified in 1981 and published in a Dutch clinical chemistry journal that highlighted a number of neurological conditions such as delayed intellectual, motor, speech, and language as the most common manifestations. Later cases reported in the early 1990s began to show that hypotonia, hyporeflexia, seizures, and a nonprogressive ataxia were frequent clinical features as well.

A 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial is an enzyme that in humans is encoded by the BCKDHA gene.

Branched chain ketoacid dehydrogenase kinase (BCKDK) is an enzyme encoded by the BCKDK gene on chromosome 16. This enzyme is part of the mitochondrial protein kinases family and it is a regulator of the valine, leucine, and isoleucine catabolic pathways. BCKDK is found in the mitochondrial matrix and the prevalence of it depends on the type of cell. Liver cells tend to have the lowest concentration of BCKDK, whereas skeletal muscle cells have the highest amount. Abnormal activity of this enzyme often leads to diseases such as maple syrup urine disease and cachexia.
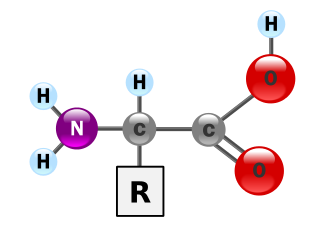
Inborn errors of amino acid metabolism are metabolic disorders which impair the synthesis and degradation of amino acids.
Nagwa Abdel Meguid is an Egyptian geneticist and 2002 winner of the L’Oreal UNESCO Award for Women in Science for Africa and the Middle East. Her research has "identified several genetic mutations that cause common syndromes such as the fragile X syndrome and Autism".

Lysine demethylase 6B is a protein that in humans is encoded by the KDM6B gene.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.
Dihydropteridine reductase deficiency (DHPRD) is a genetic disorder affecting the tetrahydrobiopterin (BH4) synthesis pathway, inherited in the autosomal recessive pattern. It is one of the six known disorders causing tetrahydrobiopterin deficiency, and occurs in patients with mutations of the QDPR gene.
CDKL5 deficiency disorder (CDD) is a rare genetic disorder caused by pathogenic variants in the gene CDKL5.
Jordan's Syndrome (JS) or PPP2R5D-related intellectual disability is a rare autosomal dominant neurodevelopmental disorder caused by de novo mutations in the PPP2R5D gene. It is characterized by hypotonia, intellectual disability, and macrocephaly. Children with JS may also have epilepsy or meet criteria for diagnosis with autism spectrum disorder.
Autosomal recessive GTP cyclohydrolase I deficiency (AR-GTPCHD) is a disorder associated with the deficient operation of the enzyme GTP cyclohydrolase I. The condition leads to insufficient production of the cofactor tetrahydrobiopterin necessary for the proper synthesis of dopamine and serotonin and for maintenance of adequate levels of phenylalanine. As of 2020, autosomal recessive GTP cyclohydrolase I deficiency was one of the six known causes of tetrahydrobiopterin deficiency. It is also considered part of the spectrum of dopa-responsive dystonias.
