
In organic chemistry, a ketene is an organic compound of the form RR'C=C=O, where R and R' are two arbitrary monovalent chemical groups. The name may also refer to the specific compound ethenone H2C=C=O, the simplest ketene.
In organic chemistry, a carbene is a molecule containing a neutral carbon atom with a valence of two and two unshared valence electrons. The general formula is R−:C−R' or R=C: where the R represents substituents or hydrogen atoms.
In organic chemistry, the diazo group is an organic moiety consisting of two linked nitrogen atoms at the terminal position. Overall charge-neutral organic compounds containing the diazo group bound to a carbon atom are called diazo compounds or diazoalkanes and are described by the general structural formula R2C=N+=N−. The simplest example of a diazo compound is diazomethane, CH2N2. Diazo compounds should not be confused with azo compounds or with diazonium compounds.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

In organic chemistry, olefin metathesis is an organic reaction that entails the redistribution of fragments of alkenes (olefins) by the scission and regeneration of carbon-carbon double bonds. Because of the relative simplicity of olefin metathesis, it often creates fewer undesired by-products and hazardous wastes than alternative organic reactions. For their elucidation of the reaction mechanism and their discovery of a variety of highly active catalysts, Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock were collectively awarded the 2005 Nobel Prize in Chemistry.

Organoboron chemistry or organoborane chemistry studies organoboron compounds, also called organoboranes. These chemical compounds combine boron and carbon; typically, they are organic derivatives of borane (BH3), as in the trialkyl boranes.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.

A persistent carbene (also known as stable carbene) is a type of carbene demonstrating particular stability. The best-known examples and by far largest subgroup are the N-heterocyclic carbenes (NHC) (sometimes called Arduengo carbenes), for example diaminocarbenes with the general formula (R2N)2C:, where the four R moieties are typically alkyl and aryl groups. The groups can be linked to give heterocyclic carbenes, such as those derived from imidazole, imidazoline, thiazole or triazole.
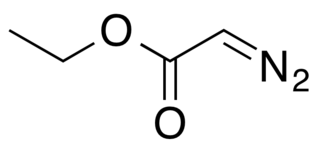
Ethyl diazoacetate (N=N=CHC(O)OC2H5) is a diazo compound and a reagent in organic chemistry. It was discovered by Theodor Curtius in 1883. The compound can be prepared by reaction of the ethyl ester of glycine with sodium nitrite and sodium acetate in water.

In organic chemistry, cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroid insecticides and a number of quinolone antibiotics. However, the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.
The triazol-5-ylidenes are a group of persistent carbenes which includes the 1,2,4-triazol-5-ylidene system and the 1,2,3-triazol-5-ylidene system. As opposed to the now ubiquitous NHC systems based on imidazole rings, these carbenes are structured from triazole rings. 1,2,4-triazol-5-ylidene can be thought of as an analog member of the NHC family, with an extra nitrogen in the ring, while 1,2,3-triazol-5-ylidene is better thought of as a mesoionic carbene (MIC). Both isomers of this group of carbenes benefit from enhanced stability, with certain examples exhibiting greater thermal stability, and others extended shelf life.
The Wanzlick equilibrium is a chemical equilibrium between a relatively stable carbene compound and its dimer. The equilibrium was proposed to apply to certain electron-rich alkenes, such as tetraminoethylenes, which have been called "carbene dimers." Such equilibria occur, but the mechanism does not proceed simply, but requires catalysts.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being collectively awarded one half of the 2001 Nobel Prize in Chemistry.

Organoruthenium chemistry is the chemistry of organometallic compounds containing a carbon to ruthenium chemical bond. Several organoruthenium catalysts are of commercial interest and organoruthenium compounds have been considered for cancer therapy. The chemistry has some stoichiometric similarities with organoiron chemistry, as iron is directly above ruthenium in group 8 of the periodic table. The most important reagents for the introduction of ruthenium are ruthenium(III) chloride and triruthenium dodecacarbonyl.
Metal-catalyzed cyclopropanations are chemical reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc. Metal carbenoid species can be generated through the reaction of a diazo compound with a transition metal). The intramolecular variant of this reaction was first reported in 1961. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are common catalysts. Enantioselective cyclopropanations have been developed.

Hydrogen auto-transfer, also known as borrowing hydrogen, is the activation of a chemical reaction by temporary transfer of two hydrogen atoms from the reactant to a catalyst and return of those hydrogen atoms back to a reaction intermediate to form the final product. Two major classes of borrowing hydrogen reactions exist: (a) those that result in hydroxyl substitution, and (b) those that result in carbonyl addition. In the former case, alcohol dehydrogenation generates a transient carbonyl compound that is subject to condensation followed by the return of hydrogen. In the latter case, alcohol dehydrogenation is followed by reductive generation of a nucleophile, which triggers carbonyl addition. As borrowing hydrogen processes avoid manipulations otherwise required for discrete alcohol oxidation and the use of stoichiometric organometallic reagents, they typically display high levels of atom-economy and, hence, are viewed as examples of Green chemistry.
A tosylhydrazone in organic chemistry is a functional group with the general structure RR'C=N-NH-Ts where Ts is a tosyl group. Organic compounds having this functional group can be accessed by reaction of an aldehyde or ketone with tosylhydrazine.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.
Carbonyl olefin metathesis is a type of metathesis reaction that entails, formally, the redistribution of fragments of an alkene and a carbonyl by the scission and regeneration of carbon-carbon and carbon-oxygen double bonds respectively. It is a powerful method in organic synthesis using simple carbonyls and olefins and converting them into less accessible products with higher structural complexity.
