An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
In chemistry, a nitrene or imene is the nitrogen analogue of a carbene. The nitrogen atom is uncharged and univalent, so it has only 6 electrons in its valence level—two covalent bonded and four non-bonded electrons. It is therefore considered an electrophile due to the unsatisfied octet. A nitrene is a reactive intermediate and is involved in many chemical reactions. The simplest nitrene, HN, is called imidogen, and that term is sometimes used as a synonym for the nitrene class.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
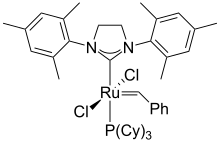
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.

A persistent carbene (also known as stable carbene) is a type of carbene demonstrating particular stability. The best-known examples and by far largest subgroup are the N-heterocyclic carbenes (NHC) (sometimes called Arduengo carbenes), for example diaminocarbenes with the general formula (R2N)2C:, where the four R moieties are typically alkyl and aryl groups. The groups can be linked to give heterocyclic carbenes, such as those derived from imidazole, imidazoline, thiazole or triazole.

Atomic carbon, systematically named carbon and λ0-methane, is a colourless gaseous inorganic chemical with the chemical formula C. It is kinetically unstable at ambient temperature and pressure, being removed through autopolymerisation.
A nitrenium ion in organic chemistry is a reactive intermediate based on nitrogen with both an electron lone pair and a positive charge and with two substituents. Nitrenium ions are isoelectronic with carbenes, and can exist in either a singlet or a triplet state. The parent nitrenium ion, NH+2, is a ground state triplet species with a gap of 30 kcal/mol (130 kJ/mol) to the lowest energy singlet state. Conversely, most arylnitrenium ions are ground state singlets. Certain substituted arylnitrenium ions can be ground state triplets, however. Nitrenium ions can have microsecond or longer lifetimes in water.
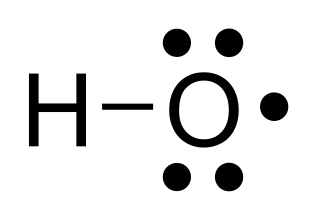
In chemistry, a radical, also known as a free radical, is an atom, molecule, or ion that has at least one unpaired valence electron. With some exceptions, these unpaired electrons make radicals highly chemically reactive. Many radicals spontaneously dimerize. Most organic radicals have short lifetimes.
In organic chemistry, diazirines are a class of organic molecules consisting of a carbon bound to two nitrogen atoms, which are double-bonded to each other, forming a cyclopropene-like ring, 3H-diazirene. They are isomeric with diazocarbon groups, and like them can serve as precursors for carbenes by loss of a molecule of dinitrogen. For example, irradiation of diazirines with ultraviolet light leads to carbene insertion into various C−H, N−H, and O−H bonds. Hence, diazirines have grown in popularity as small, photo-reactive, crosslinking reagents. They are often used in photoaffinity labeling studies to observe a variety of interactions, including ligand-receptor, ligand-enzyme, protein-protein, and protein-nucleic acid interactions.

Carbene C−H insertion in organic chemistry concerns the insertion reaction of a carbene into a carbon–hydrogen bond. This organic reaction is of some importance in the synthesis of new organic compounds.
Carbene analogs in chemistry are carbenes with the carbon atom replaced by another chemical element. Just as regular carbenes they appear in chemical reactions as reactive intermediates and with special precautions they can be stabilized and isolated as chemical compounds. Carbenes have some practical utility in organic synthesis but carbene analogs are mostly laboratory curiosities only investigated in academia. Carbene analogs are known for elements of group 13, group 14, group 15 and group 16.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.

Phosphinidenes are low-valent phosphorus compounds analogous to carbenes and nitrenes, having the general structure RP. The "free" form of these compounds is conventionally described as having a singly-coordinated phosphorus atom containing only 6 electrons in its valence level. Most phosphinidenes are highly reactive and short-lived, thereby complicating empirical studies on their chemical properties. In the last few decades, several strategies have been employed to stabilize phosphinidenes, and researchers have developed a number of reagents and systems that can generate and transfer phosphinidenes as reactive intermediates in the synthesis of various organophosphorus compounds.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
Methylene is an organic compound with the chemical formula CH
2. It is a colourless gas that fluoresces in the mid-infrared range, and only persists in dilution, or as an adduct.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.

9-Fluorenylidene is an aryl carbene derived from the bridging methylene group of fluorene. Fluorenylidene has the unusual property that the triplet ground state is only 1.1 kcal/mol lower in energy than the singlet state. For this reason, fluorenylidene has been studied extensively in organic chemistry.
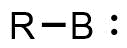
A borylene is the boron analogue of a carbene. The general structure is R-B: with R an organic moiety and B a boron atom with two unshared electrons. Borylenes are of academic interest in organoboron chemistry. A singlet ground state is predominant with boron having two vacant sp2 orbitals and one doubly occupied one. With just one additional substituent the boron is more electron deficient than the carbon atom in a carbene. For this reason stable borylenes are more uncommon than stable carbenes. Some borylenes such as boron monofluoride (BF) and boron monohydride (BH) the parent compound also known simply as borylene, have been detected in microwave spectroscopy and may exist in stars. Other borylenes exist as reactive intermediates and can only be inferred by chemical trapping.

Plumbylenes (or plumbylidenes) are divalent organolead(II) analogues of carbenes, with the general chemical formula, R2Pb, where R denotes a substituent. Plumbylenes possess 6 electrons in their valence shell, and are considered open shell species.
A Fischer carbene is a type of transition metal carbene complex, which is an organometallic compound containing a divalent organic ligand. In a Fischer carbene, the carbene ligand is a σ-donor π-acceptor ligand. Because π-backdonation from the metal centre is generally weak, the carbene carbon is electrophilic.