A chemical bond is a lasting attraction between atoms or ions that enables the formation of molecules, crystals, and other structures. The bond may result from the electrostatic force between oppositely charged ions as in ionic bonds, or through the sharing of electrons as in covalent bonds. The strength of chemical bonds varies considerably: there are "strong bonds" or "primary bonds" such as covalent, ionic and metallic bonds, and "weak bonds" or "secondary bonds" such as dipole–dipole interactions, the London dispersion force, and hydrogen bonding.

A covalent bond is a chemical bond that involves the sharing of electrons to form electron pairs between atoms. These electron pairs are known as shared pairs or bonding pairs. The stable balance of attractive and repulsive forces between atoms, when they share electrons, is known as covalent bonding. For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full valence shell, corresponding to a stable electronic configuration. In organic chemistry, covalent bonding is much more common than ionic bonding.
An intermolecular force (IMF) is the force that mediates interaction between molecules, including the electromagnetic forces of attraction or repulsion which act between atoms and other types of neighbouring particles, e.g. atoms or ions. Intermolecular forces are weak relative to intramolecular forces – the forces which hold a molecule together. For example, the covalent bond, involving sharing electron pairs between atoms, is much stronger than the forces present between neighboring molecules. Both sets of forces are essential parts of force fields frequently used in molecular mechanics.

In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.

In chemistry, polarity is a separation of electric charge leading to a molecule or its chemical groups having an electric dipole moment, with a negatively charged end and a positively charged end.

Valence shell electron pair repulsion (VSEPR) theory is a model used in chemistry to predict the geometry of individual molecules from the number of electron pairs surrounding their central atoms. It is also named the Gillespie-Nyholm theory after its two main developers, Ronald Gillespie and Ronald Nyholm.
Ligand field theory (LFT) describes the bonding, orbital arrangement, and other characteristics of coordination complexes. It represents an application of molecular orbital theory to transition metal complexes. A transition metal ion has nine valence atomic orbitals - consisting of five nd, one (n+1)s, and three (n+1)p orbitals. These orbitals have the appropriate energy to form bonding interactions with ligands. The LFT analysis is highly dependent on the geometry of the complex, but most explanations begin by describing octahedral complexes, where six ligands coordinate with the metal. Other complexes can be described with reference to crystal field theory. Inverted ligand field theory (ILFT) elaborates on LFT by breaking assumptions made about relative metal and ligand orbital energies.

In chemistry, π backbonding, also called π backdonation, is when electrons move from an atomic orbital on one atom to an appropriate symmetry antibonding orbital on a π-acceptor ligand. It is especially common in the organometallic chemistry of transition metals with multi-atomic ligands such as carbon monoxide, ethylene or the nitrosonium cation. Electrons from the metal are used to bond to the ligand, in the process relieving the metal of excess negative charge. Compounds where π backbonding occurs include Ni(CO)4 and Zeise's salt. IUPAC offers the following definition for backbonding:
A description of the bonding of π-conjugated ligands to a transition metal which involves a synergic process with donation of electrons from the filled π-orbital or lone electron pair orbital of the ligand into an empty orbital of the metal (donor–acceptor bond), together with release (back donation) of electrons from an nd orbital of the metal (which is of π-symmetry with respect to the metal–ligand axis) into the empty π*-antibonding orbital of the ligand.
In chemistry, orbital hybridisation is the concept of mixing atomic orbitals to form new hybrid orbitals suitable for the pairing of electrons to form chemical bonds in valence bond theory. For example, in a carbon atom which forms four single bonds the valence-shell s orbital combines with three valence-shell p orbitals to form four equivalent sp3 mixtures in a tetrahedral arrangement around the carbon to bond to four different atoms. Hybrid orbitals are useful in the explanation of molecular geometry and atomic bonding properties and are symmetrically disposed in space. Usually hybrid orbitals are formed by mixing atomic orbitals of comparable energies.

In chemistry, a trigonal pyramid is a molecular geometry with one atom at the apex and three atoms at the corners of a trigonal base, resembling a tetrahedron (not to be confused with the tetrahedral geometry). When all three atoms at the corners are identical, the molecule belongs to point group C3v. Some molecules and ions with trigonal pyramidal geometry are the pnictogen hydrides (XH3), xenon trioxide (XeO3), the chlorate ion, ClO−
3, and the sulfite ion, SO2−
3. In organic chemistry, molecules which have a trigonal pyramidal geometry are sometimes described as sp3 hybridized. The AXE method for VSEPR theory states that the classification is AX3E1.
In chemistry, a non-covalent interaction differs from a covalent bond in that it does not involve the sharing of electrons, but rather involves more dispersed variations of electromagnetic interactions between molecules or within a molecule. The chemical energy released in the formation of non-covalent interactions is typically on the order of 1–5 kcal/mol. Non-covalent interactions can be classified into different categories, such as electrostatic, π-effects, van der Waals forces, and hydrophobic effects.
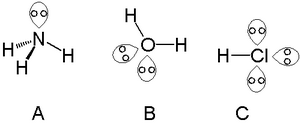
In chemistry, Bent's rule describes and explains the relationship between the orbital hybridization and the electronegativities of substituents. The rule was stated by Henry A. Bent as follows:
Atomic s character concentrates in orbitals directed toward electropositive substituents.
A molecular orbital diagram, or MO diagram, is a qualitative descriptive tool explaining chemical bonding in molecules in terms of molecular orbital theory in general and the linear combination of atomic orbitals (LCAO) method in particular. A fundamental principle of these theories is that as atoms bond to form molecules, a certain number of atomic orbitals combine to form the same number of molecular orbitals, although the electrons involved may be redistributed among the orbitals. This tool is very well suited for simple diatomic molecules such as dihydrogen, dioxygen, and carbon monoxide but becomes more complex when discussing even comparatively simple polyatomic molecules, such as methane. MO diagrams can explain why some molecules exist and others do not. They can also predict bond strength, as well as the electronic transitions that can take place.
Localized molecular orbitals are molecular orbitals which are concentrated in a limited spatial region of a molecule, such as a specific bond or lone pair on a specific atom. They can be used to relate molecular orbital calculations to simple bonding theories, and also to speed up post-Hartree–Fock electronic structure calculations by taking advantage of the local nature of electron correlation. Localized orbitals in systems with periodic boundary conditions are known as Wannier functions.

The linear molecular geometry describes the geometry around a central atom bonded to two other atoms placed at a bond angle of 180°. Linear organic molecules, such as acetylene, are often described by invoking sp orbital hybridization for their carbon centers.
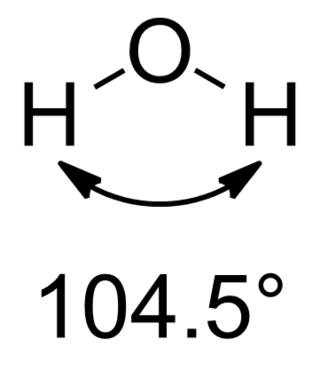
In chemistry, molecules with a non-collinear arrangement of two adjacent bonds have bent molecular geometry, also known as angular or V-shaped. Certain atoms, such as oxygen, will almost always set their two (or more) covalent bonds in non-collinear directions due to their electron configuration. Water (H2O) is an example of a bent molecule, as well as its analogues. The bond angle between the two hydrogen atoms is approximately 104.45°. Nonlinear geometry is commonly observed for other triatomic molecules and ions containing only main group elements, prominent examples being nitrogen dioxide (NO2), sulfur dichloride (SCl2), and methylene (CH2).
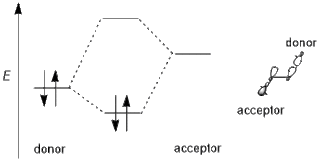
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
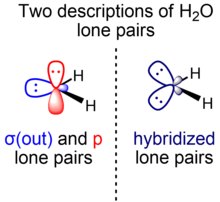
The σ-π model and equivalent-orbital model refer to two possible representations of molecules in valence bond theory. The σ-π model differentiates bonds and lone pairs of σ symmetry from those of π symmetry, while the equivalent-orbital model hybridizes them. The σ-π treatment takes into account molecular symmetry and is better suited to interpretation of aromatic molecules, although computational calculations of certain molecules tend to optimize better under the equivalent-orbital treatment. The two representations produce the same total electron density and are related by a unitary transformation of the occupied molecular orbitals; different localization procedures yield either of the two. Two equivalent orbitals h and h' can be constructed by taking linear combinations h = c1σ + c2π and h' = c1σ – c2π for an appropriate choice of coefficients c1 and c2.

Water is a simple triatomic bent molecule with C2v molecular symmetry and bond angle of 104.5° between the central oxygen atom and the hydrogen atoms. Despite being one of the simplest triatomic molecules, its chemical bonding scheme is nonetheless complex as many of its bonding properties such as bond angle, ionization energy, and electronic state energy cannot be explained by one unified bonding model. Instead, several traditional and advanced bonding models such as simple Lewis and VSEPR structure, valence bond theory, molecular orbital theory, isovalent hybridization, and Bent's rule are discussed below to provide a comprehensive bonding model for H
2O, explaining and rationalizing the various electronic and physical properties and features manifested by its peculiar bonding arrangements.
Digermynes are a class of compounds that are regarded as the heavier digermanium analogues of alkynes. The parent member of this entire class is HGeGeH, which has only been characterized computationally, but has revealed key features of the whole class. Because of the large interatomic repulsion between two Ge atoms, only kinetically stabilized digermyne molecules can be synthesized and characterized by utilizing bulky protecting groups and appropriate synthetic methods, for example, reductive coupling of germanium(II) halides.