Related Research Articles

Neurofibromatosis (NF) is a group of three conditions in which tumors grow in the nervous system. The three types are neurofibromatosis type I (NF1), neurofibromatosis type II (NF2), and schwannomatosis. In NF1 symptoms include light brown spots on the skin, freckles in the armpit and groin, small bumps within nerves, and scoliosis. In NF2, there may be hearing loss, cataracts at a young age, balance problems, flesh colored skin flaps, and muscle wasting. In schwannomatosis there may be pain either in one location or in wide areas of the body. The tumors in NF are generally non-cancerous.

Hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome is an autosomal dominant genetic condition that is associated with a high risk of colon cancer as well as other cancers including endometrial cancer, ovary, stomach, small intestine, hepatobiliary tract, upper urinary tract, brain, and skin. The increased risk for these cancers is due to inherited mutations that impair DNA mismatch repair. It is a type of cancer syndrome. Because patients with Lynch syndrome can have polyps, the term HNPCC has fallen out of favor.

Café au lait spots, or café au lait macules, are flat, hyperpigmented birthmarks. The name café au lait is French for "coffee with milk" and refers to their light-brown color. Café au lait lesions with rough borders may be seen in McCune-Albright syndrome. In contrast, café au lait lesions of neurofibromatosis have smooth borders.

Mismatch repair cancer syndrome (MMRCS) is a cancer syndrome associated with biallelic DNA mismatch repair mutations. It is also known as Turcot syndrome and by several other names.

Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of neurofibromin, a gene on chromosome 17 that is responsible for production of a protein which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.
In genetics, expressivity is the degree to which a phenotype is expressed by individuals having a particular genotype. Expressivity is related to the intensity of a given phenotype; it differs from penetrance, which refers to the proportion of individuals with a particular genotype that actually express the phenotype.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s), in which multiple cranial nerves can be involved. Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.
The oral mucosa is the mucous membrane lining the inside of the mouth. It comprises stratified squamous epithelium, termed "oral epithelium", and an underlying connective tissue termed lamina propria. The oral cavity has sometimes been described as a mirror that reflects the health of the individual. Changes indicative of disease are seen as alterations in the oral mucosa lining the mouth, which can reveal systemic conditions, such as diabetes or vitamin deficiency, or the local effects of chronic tobacco or alcohol use. The oral mucosa tends to heal faster and with less scar formation compared to the skin. The underlying mechanism remains unknown, but research suggests that extracellular vesicles might be involved.

A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
Phakomatoses, also known neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.

McCune–Albright syndrome is a complex genetic disorder affecting the bone, skin and endocrine systems. It is a mosaic disease arising from somatic activating mutations in GNAS, which encodes the alpha-subunit of the Gs heterotrimeric G protein.

Noonan syndrome with multiple lentigines (NSML) which is part of a group called Ras/MAPK pathway syndromes, is a rare autosomal dominant, multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.

Neurofibromin 1 (NF1) is a gene in humans that is located on chromosome 17. NF1 codes for neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations in NF1 can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.
Juvenile myelomonocytic leukemia (JMML) is a rare and serious chronic leukemia (cancer of the blood) that affects children mostly aged 4 and younger. The name JMML now encompasses all diagnoses formerly referred to as juvenile chronic myeloid leukemia (JCML), chronic myelomonocytic leukemia of infancy, and infantile monosomy 7 syndrome. The average age of patients at diagnosis is 2 years old. The World Health Organization has included JMML in the category of myelodysplastic and myeloproliferative disorders.
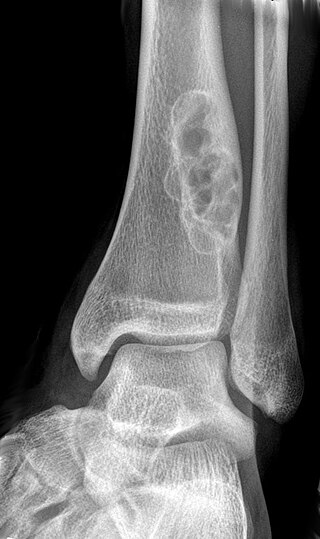
A non-ossifying fibroma (NOF) is a benign bone tumor of the osteoclastic giant cell-rich tumor type. It generally occurs in the metaphysis of long bones in children and adolescents. Typically, there are no symptoms unless there is a fracture. It can occur as part of a syndrome such as when multiple non-ossifying fibromas occur in neurofibromatosis, or Jaffe-Campanacci syndrome in combination with cafe-au-lait spots, mental retardation, hypogonadism, eye and cardiovascular abnormalities.
Frank Wilkinson Crowe was an American physician, who practiced Dermatology in Boise, Idaho. He was a world-renowned authority for the Crowe sign or Crowe's sign in neurofibromatosis which is the presence of axillary (armpit) freckling in people with neurofibromatosis type I. These freckles occur in up to 30% of people with the disease and their presence is one of seven diagnostic criteria for neurofibromatosis. Freckles can also be present in the intertriginous area in neurofibromatosis, such as the inguinal fold, submamillary areas and nape of the neck.

Palisaded encapsulated neuroma (PEN) is a rare, benign cutaneous condition characterized by small, firm, non-pigmented nodules or papules. They typically occur as a solitary (single) lesion near the mucocutaneous junction of the skin of the face, although they can occur elsewhere on the body.

Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I (NF-1). It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome (NFLS).

Ring chromosome 15 is a condition that arises when chromosome 15 fuses to form a ring chromosome. Usually, ring chromosome 15 forms due to the modification or deletion of genetic information on chromosome 15 in the preliminary stages of embryonic development, but it can rarely also be inherited.
Familial multiple cafe au lait spots, also known as Autosomal dominant multiple cafe au lait spots or Neurofibromatosis type 6, is a rare, cutaneous genetic disorder which is characterized by the hereditary cutaneous presence of several cafe-au-lait spots without any other symptoms of neurofibromatosis. Sporadic cases may be called "Sporadic multiple cafe au lait spots". Few cases have been described in medical literature, although it is estimated that the presence of multiple cafe au lait spots without NF1 is rare among the general population.
References
- ↑ Crowe, Frank (1956). A clinical, pathological and genetic study of multiple neurofibromatosis. Thomas, Charles C. Publisher, Ltd. ISBN 9780398003708.
- ↑ K Jett, JM Friedman. Clinical and Genetic Aspects of Neurofibromatosis 1. Genetics in Medicine 12(1):1-11. January 2010.
| | This dermatology article is a stub. You can help Wikipedia by expanding it. |