Pyruvate dehydrogenase complex (PDC) is a complex of three enzymes that converts pyruvate into acetyl-CoA by a process called pyruvate decarboxylation. Acetyl-CoA may then be used in the citric acid cycle to carry out cellular respiration, and this complex links the glycolysis metabolic pathway to the citric acid cycle. Pyruvate decarboxylation is also known as the "pyruvate dehydrogenase reaction" because it also involves the oxidation of pyruvate.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness. It was described by John Menkes in the 1950s.
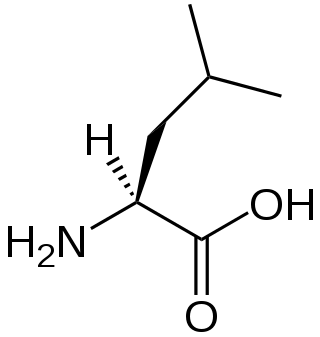
A branched-chain amino acid (BCAA) is an amino acid having an aliphatic side-chain with a branch. Among the proteinogenic amino acids, there are three BCAAs: leucine, isoleucine, and valine. Non-proteinogenic BCAAs include 2-aminoisobutyric acid and alloisoleucine.
The branched-chain α-ketoacid dehydrogenase complex is a multi-subunit complex of enzymes that is found on the mitochondrial inner membrane. This enzyme complex catalyzes the oxidative decarboxylation of branched, short-chain alpha-ketoacids. BCKDC is a member of the mitochondrial α-ketoacid dehydrogenase complex family, which also includes pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, key enzymes that function in the Krebs cycle.
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.

Dihydrolipoyl transacetylase is an enzyme component of the multienzyme pyruvate dehydrogenase complex. The pyruvate dehydrogenase complex is responsible for the pyruvate decarboxylation step that links glycolysis to the citric acid cycle. This involves the transformation of pyruvate from glycolysis into acetyl-CoA which is then used in the citric acid cycle to carry out cellular respiration.

Dihydrolipoamide dehydrogenase (DLD), also known as dihydrolipoyl dehydrogenase, mitochondrial, is an enzyme that in humans is encoded by the DLD gene. DLD is a flavoprotein enzyme that oxidizes dihydrolipoamide to lipoamide.
E3 binding protein also known as pyruvate dehydrogenase protein X component, mitochondrial is a protein that in humans is encoded by the PDHX gene. The E3 binding protein is a component of the pyruvate dehydrogenase complex found only in eukaryotes. Defects in this gene are a cause of pyruvate dehydrogenase deficiency which results in neurological dysfunction and lactic acidosis in infancy and early childhood. This protein is also a minor antigen for antimitochondrial antibodies. These autoantibodies are present in nearly 95% of patients with primary biliary cholangitis, an autoimmune disease of the liver. In primary biliary cholangitis, activated T lymphocytes attack and destroy epithelial cells in the bile duct where this protein is abnormally distributed and overexpressed. Primary biliary cholangitis eventually leads to liver failure.

In enzymology, a malate dehydrogenase (NADP+) (EC 1.1.1.82) is an enzyme that catalyzes the chemical reaction

In enzymology, a 3-methyl-2-oxobutanoate dehydrogenase (EC 1.2.4.4) is an enzyme that catalyzes the chemical reaction

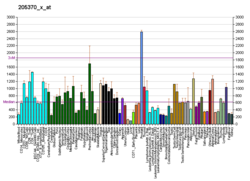
Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

In enzymology, a dihydrolipoyllysine-residue (2-methylpropanoyl)transferase (EC 2.3.1.168) is an enzyme that catalyzes the chemical reaction
In enzymology, a [3-methyl-2-oxobutanoate dehydrogenase (acetyl-transferring)] is an enzyme that catalyzes the chemical reaction

Aldo-keto reductase family 1 member C1 also known as 20α-hydroxysteroid dehydrogenase, 3α-hydroxysteroid dehydrogenase, and dihydrodiol dehydrogenase 1/2 is an enzyme that in humans is encoded by the AKR1C1 gene.

A 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial is an enzyme that in humans is encoded by the BCKDHA gene.

Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial is an enzyme that in humans is encoded by the DLST gene.

2-Oxoisovalerate dehydrogenase subunit beta, mitochondrial is an enzyme that in humans is encoded by the BCKDHB gene.

Pyruvate dehydrogenase lipoamide kinase isozyme 3, mitochondrial is an enzyme that in humans is encoded by the PDK3 gene. It codes for an isozyme of pyruvate dehydrogenase kinase.The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2. It provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle, and thus is one of the major enzymes responsible for the regulation of glucose metabolism. The enzymatic activity of PDH is regulated by a phosphorylation/dephosphorylation cycle, and phosphorylation results in inactivation of PDH. The protein encoded by this gene is one of the four pyruvate dehydrogenase kinases that inhibits the PDH complex by phosphorylation of the E1 alpha subunit. This gene is predominantly expressed in the heart and skeletal muscles. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.

Branched chain ketoacid dehydrogenase kinase (BCKDK) is an enzyme encoded by the BCKDK gene on chromosome 16. This enzyme is part of the mitochondrial protein kinases family and it is a regulator of the valine, leucine, and isoleucine catabolic pathways. BCKDK is found in the mitochondrial matrix and the prevalence of it depends on the type of cell. Liver cells tend to have the lowest concentration of BCKDK, whereas skeletal muscle cells have the highest amount. Abnormal activity of this enzyme often leads to diseases such as maple syrup urine disease and cachexia.

α-Ketoisocaproic acid (α-KIC), also known as 4-methyl-2-oxovaleric acid, and its conjugate base and carboxylate, α-ketoisocaproate, are metabolic intermediates in the metabolic pathway for L-leucine. Leucine is an essential amino acid, and its degradation is critical for many biological duties. α-KIC is produced in one of the first steps of the pathway by branched-chain amino acid aminotransferase by transferring the amine on L-leucine onto alpha ketoglutarate, and replacing that amine with a ketone. The degradation of L-leucine in the muscle to this compound allows for the production of the amino acids alanine and glutamate as well. In the liver, α-KIC can be converted to a vast number of compounds depending on the enzymes and cofactors present, including cholesterol, acetyl-CoA, isovaleryl-CoA, and other biological molecules. Isovaleryl-CoA is the main compound synthesized from ɑ-KIC. α-KIC is a key metabolite present in the urine of people with Maple syrup urine disease, along with other branched-chain amino acids. Derivatives of α-KIC have been studied in humans for their ability to improve physical performance during anaerobic exercise as a supplemental bridge between short-term and long-term exercise supplements. These studies show that α-KIC does not achieve this goal without other ergogenicsupplements present as well. α-KIC has also been observed to reduce skeletal muscle damage after eccentrically biased resistance exercises in people who do not usually perform those exercises.