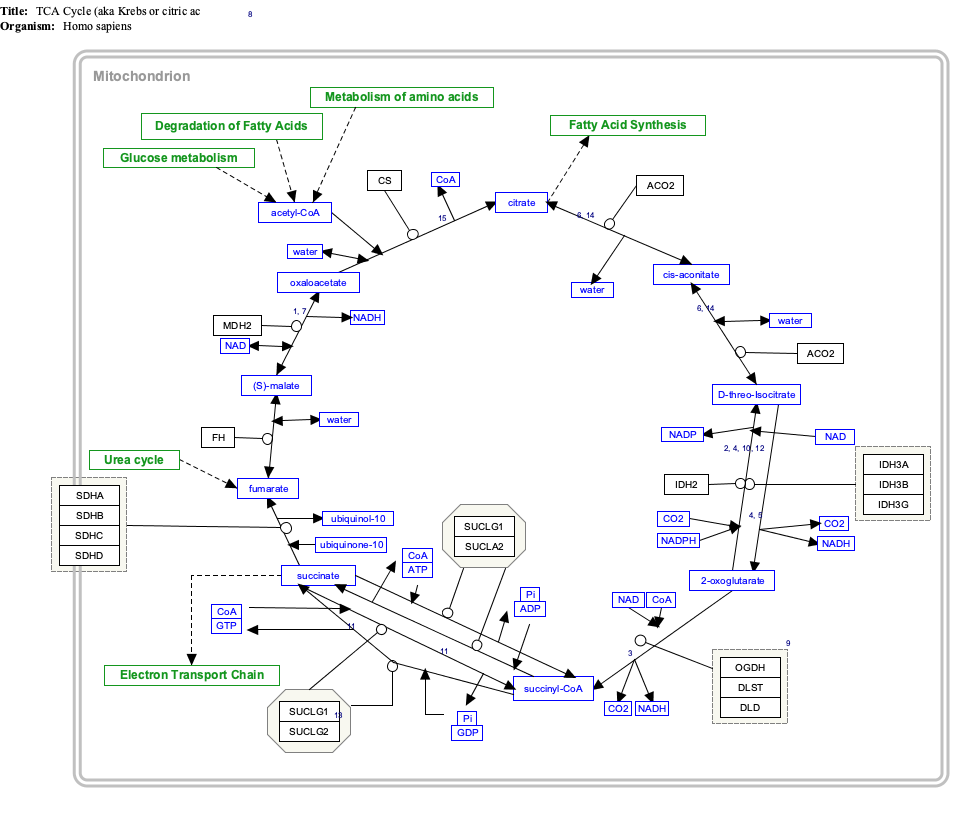
The citric acid cycle —also known as the Krebs cycle, Szent-Györgyi-Krebs cycle or the TCA cycle (tricarboxylic acid cycle)—is a series of chemical reactions to release stored energy through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins. The Krebs cycle is used by organisms that respire (as opposed to organisms that ferment) to generate energy, either by anaerobic respiration or aerobic respiration. In addition, the cycle provides precursors of certain amino acids, as well as the reducing agent NADH, that are used in numerous other reactions. Its central importance to many biochemical pathways suggests that it was one of the earliest components of metabolism. Even though it is branded as a 'cycle', it is not necessary for metabolites to follow only one specific route; at least three alternative segments of the citric acid cycle have been recognized.

Pyruvate dehydrogenase complex (PDC) is a complex of three enzymes that converts pyruvate into acetyl-CoA by a process called pyruvate decarboxylation. Acetyl-CoA may then be used in the citric acid cycle to carry out cellular respiration, and this complex links the glycolysis metabolic pathway to the citric acid cycle. Pyruvate decarboxylation is also known as the "pyruvate dehydrogenase reaction" because it also involves the oxidation of pyruvate.
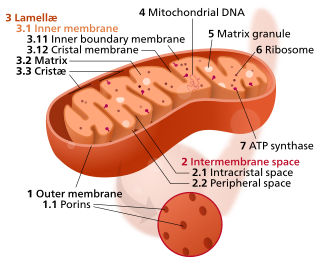
In the mitochondrion, the matrix is the space within the inner membrane. The word "matrix" stems from the fact that this space is viscous, compared to the relatively aqueous cytoplasm. The mitochondrial matrix contains the mitochondrial DNA, ribosomes, soluble enzymes, small organic molecules, nucleotide cofactors, and inorganic ions.[1] The enzymes in the matrix facilitate reactions responsible for the production of ATP, such as the citric acid cycle, oxidative phosphorylation, oxidation of pyruvate, and the beta oxidation of fatty acids.
Pyruvate dehydrogenase lipoamide kinase isozyme 1, mitochondrial is an enzyme that in humans is encoded by the PDK1 gene. It codes for an isozyme of pyruvate dehydrogenase kinase (PDK).
The branched-chain α-ketoacid dehydrogenase complex is a multi-subunit complex of enzymes that is found on the mitochondrial inner membrane. This enzyme complex catalyzes the oxidative decarboxylation of branched, short-chain alpha-ketoacids. BCKDC is a member of the mitochondrial α-ketoacid dehydrogenase complex family comprising pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, key enzymes that function in the Krebs cycle.
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.

Dihydrolipoyl transacetylase is an enzyme component of the multienzyme pyruvate dehydrogenase complex. The pyruvate dehydrogenase complex is responsible for the pyruvate decarboxylation step that links glycolysis to the citric acid cycle. This involves the transformation of pyruvate from glycolysis into acetyl-CoA which is then used in the citric acid cycle to carry out cellular respiration.
Oxidative decarboxylation is a decarboxylation reaction caused by oxidation. Most are accompanied by α- Ketoglutarate α- Decarboxylation caused by dehydrogenation of hydroxyl carboxylic acids such as carbonyl carboxylic acid, malic acid, isocitric acid, etc.
Pyruvate dehydrogenase is an enzyme that catalyzes the reaction of pyruvate and a lipoamide to give the acetylated dihydrolipoamide and carbon dioxide. The conversion requires the coenzyme thiamine pyrophosphate.

Pyruvate dehydrogenase kinase is a kinase enzyme which acts to inactivate the enzyme pyruvate dehydrogenase by phosphorylating it using ATP.

E3 binding protein also known as pyruvate dehydrogenase protein X component, mitochondrial is a protein that in humans is encoded by the PDHX gene. The E3 binding protein is a component of the pyruvate dehydrogenase complex found only in eukaryotes. Defects in this gene are a cause of pyruvate dehydrogenase deficiency which results in neurological dysfunction and lactic acidosis in infancy and early childhood. This protein is also a minor antigen for antimitochondrial antibodies. These autoantibodies are present in nearly 95% of patients with primary biliary cholangitis, an autoimmune disease of the liver. In primary biliary cholangitis, activated T lymphocytes attack and destroy epithelial cells in the bile duct where this protein is abnormally distributed and overexpressed. Primary biliary cholangitis eventually leads to liver failure.

Glycerol-3-phosphate dehydrogenase (GPDH) is an enzyme that catalyzes the reversible redox conversion of dihydroxyacetone phosphate to sn-glycerol 3-phosphate.

In enzymology, a glycerate dehydrogenase (EC 1.1.1.29) is an enzyme that catalyzes the chemical reaction

Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

Lipoamide acyltransferase component of branched-chain alpha-keto acid dehydrogenase complex, mitochondrial is an enzyme that in humans is encoded by the DBT gene.

Pyruvate dehydrogenase kinase isoform 2 (PDK2) also known as pyruvate dehydrogenase lipoamide kinase isozyme 2, mitochondrial is an enzyme that in humans is encoded by the PDK2 gene. PDK2 is an isozyme of pyruvate dehydrogenase kinase.

Pyruvate dehydrogenase lipoamide kinase isozyme 3, mitochondrial is an enzyme that in humans is encoded by the PDK3 gene. It codes for an isozyme of pyruvate dehydrogenase kinase.The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2. It provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle, and thus is one of the major enzymes responsible for the regulation of glucose metabolism. The enzymatic activity of PDH is regulated by a phosphorylation/dephosphorylation cycle, and phosphorylation results in inactivation of PDH. The protein encoded by this gene is one of the four pyruvate dehydrogenase kinases that inhibits the PDH complex by phosphorylation of the E1 alpha subunit. This gene is predominantly expressed in the heart and skeletal muscles. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.

NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7, also known as complex I-B18, is an enzyme that in humans is encoded by the NDUFB7 gene. NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 7 is an accessory subunit of the NADH dehydrogenase (ubiquinone) complex, located in the mitochondrial inner membrane. It is also known as Complex I and is the largest of the five complexes of the electron transport chain.

Pyruvate dehydrogenase (lipoamide) alpha 2, also known as pyruvate dehydrogenase E1 component subunit alpha, testis-specific form, mitochondrial or PDHE1-A type II, is an enzyme that in humans is encoded by the PDHA2 gene.

Pyruvate dehydrogenase (lipoamide) beta, also known as pyruvate dehydrogenase E1 component subunit beta, mitochondrial or PDHE1-B is an enzyme that in humans is encoded by the PDHB gene. The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2, and provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle. The PDH complex is composed of multiple copies of three enzymatic components: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2) and lipoamide dehydrogenase (E3). The E1 enzyme is a heterotetramer of two alpha and two beta subunits. This gene encodes the E1 beta subunit. Mutations in this gene are associated with pyruvate dehydrogenase E1-beta deficiency.