
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

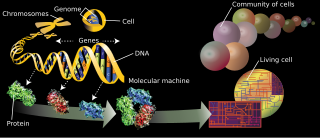
Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.

In population genetics, gene flow is the transfer of genetic material from one population to another. If the rate of gene flow is high enough, then two populations will have equivalent allele frequencies and therefore can be considered a single effective population. It has been shown that it takes only "one migrant per generation" to prevent populations from diverging due to drift. Populations can diverge due to selection even when they are exchanging alleles, if the selection pressure is strong enough. Gene flow is an important mechanism for transferring genetic diversity among populations. Migrants change the distribution of genetic diversity among populations, by modifying allele frequencies. High rates of gene flow can reduce the genetic differentiation between the two groups, increasing homogeneity. For this reason, gene flow has been thought to constrain speciation and prevent range expansion by combining the gene pools of the groups, thus preventing the development of differences in genetic variation that would have led to differentiation and adaptation. In some cases dispersal resulting in gene flow may also result in the addition of novel genetic variants under positive selection to the gene pool of a species or population
The branches of science known informally as omics are various disciplines in biology whose names end in the suffix -omics, such as genomics, proteomics, metabolomics, metagenomics, phenomics and transcriptomics. Omics aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism or organisms.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.
Conservation genetics is an interdisciplinary subfield of population genetics that aims to understand the dynamics of genes in a population for the purpose of natural resource management and extinction prevention. Researchers involved in conservation genetics come from a variety of fields including population genetics, natural resources, molecular ecology, biology, evolutionary biology, and systematics. Genetic diversity is one of the three fundamental measures of biodiversity, so it is an important consideration in the wider field of conservation biology.

Molecular ecology is a field of evolutionary biology that is concerned with applying molecular population genetics, molecular phylogenetics, and more recently genomics to traditional ecological questions. It is virtually synonymous with the field of "Ecological Genetics" as pioneered by Theodosius Dobzhansky, E. B. Ford, Godfrey M. Hewitt, and others. These fields are united in their attempt to study genetic-based questions "out in the field" as opposed to the laboratory. Molecular ecology is related to the field of conservation genetics.
In genetics, the term synteny refers to two related concepts:
Inbreeding depression is the reduced biological fitness which has the potential to result from inbreeding. Biological fitness refers to an organism's ability to survive and perpetuate its genetic material. Inbreeding depression is often the result of a population bottleneck. In general, the higher the genetic variation or gene pool within a breeding population, the less likely it is to suffer from inbreeding depression, though inbreeding and outbreeding depression can simultaneously occur.
Coalescent theory is a model of how alleles sampled from a population may have originated from a common ancestor. In the simplest case, coalescent theory assumes no recombination, no natural selection, and no gene flow or population structure, meaning that each variant is equally likely to have been passed from one generation to the next. The model looks backward in time, merging alleles into a single ancestral copy according to a random process in coalescence events. Under this model, the expected time between successive coalescence events increases almost exponentially back in time. Variance in the model comes from both the random passing of alleles from one generation to the next, and the random occurrence of mutations in these alleles.

The Human Genome Project (HGP) was an international scientific research project with the goal of determining the base pairs that make up human DNA, and of identifying, mapping and sequencing all of the genes of the human genome from both a physical and a functional standpoint. It started in 1990 and was completed in 2003. It remains the world's largest collaborative biological project. Planning for the project started after it was adopted in 1984 by the US government, and it officially launched in 1990. It was declared complete on April 14, 2003, and included about 92% of the genome. Level "complete genome" was achieved in May 2021, with a remaining only 0.3% bases covered by potential issues. The final gapless assembly was finished in January 2022.
Human evolutionary genetics studies how one human genome differs from another human genome, the evolutionary past that gave rise to the human genome, and its current effects. Differences between genomes have anthropological, medical, historical and forensic implications and applications. Genetic data can provide important insights into human evolution.
Population genomics is the large-scale comparison of DNA sequences of populations. Population genomics is a neologism that is associated with population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.

A reference genome is a digital nucleic acid sequence database, assembled by scientists as a representative example of the set of genes in one idealized individual organism of a species. As they are assembled from the sequencing of DNA from a number of individual donors, reference genomes do not accurately represent the set of genes of any single individual organism. Instead, a reference provides a haploid mosaic of different DNA sequences from each donor. For example, one of the most recent human reference genomes, assembly GRCh38/hg38, is derived from >60 genomic clone libraries. There are reference genomes for multiple species of viruses, bacteria, fungus, plants, and animals. Reference genomes are typically used as a guide on which new genomes are built, enabling them to be assembled much more quickly and cheaply than the initial Human Genome Project. Reference genomes can be accessed online at several locations, using dedicated browsers such as Ensembl or UCSC Genome Browser.
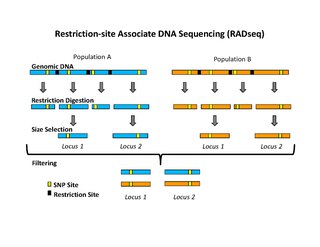
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.

Landscape genetics is the scientific discipline that combines population genetics and landscape ecology. It broadly encompasses any study that analyses plant or animal population genetic data in conjunction with data on the landscape features and matrix quality where the sampled population lives. This allows for the analysis of microevolutionary processes affecting the species in light of landscape spatial patterns, providing a more realistic view of how populations interact with their environments. Landscape genetics attempts to determine which landscape features are barriers to dispersal and gene flow, how human-induced landscape changes affect the evolution of populations, the source-sink dynamics of a given population, and how diseases or invasive species spread across landscapes.

