
Methylmalonic acidemia, also called methylmalonic aciduria, is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism. It is a classical type of organic acidemia. The result of this condition is the inability to properly digest specific fats and proteins, which in turn leads to a buildup of a toxic level of methylmalonic acid in the blood.

Methionine synthase (MS, MeSe, MTR) is responsible for the regeneration of methionine from homocysteine. In humans it is encoded by the MTR gene (5-methyltetrahydrofolate-homocysteine methyltransferase). Methionine synthase forms part of the S-adenosylmethionine (SAMe) biosynthesis and regeneration cycle, and is the enzyme responsible for linking the cycle to one-carbon metabolism via the folate cycle. There are two primary forms of this enzyme, the Vitamin B12 (cobalamin)-dependent (MetH) and independent (MetE) forms, although minimal core methionine synthases that do not fit cleanly into either category have also been described in some anaerobic bacteria. The two dominant forms of the enzymes appear to be evolutionary independent and rely on considerably different chemical mechanisms. Mammals and other higher eukaryotes express only the cobalamin-dependent form. In contrast, the distribution of the two forms in Archaeplastida (plants and algae) is more complex. Plants exclusively possess the cobalamin-independent form, while algae have either one of the two, depending on species. Many different microorganisms express both the cobalamin-dependent and cobalamin-independent forms.

Malonic aciduria or malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.

Methylmalonyl-CoA mutase is a mitochondrial homodimer apoenzyme that focuses on the catalysis of methylmalonyl CoA to succinyl CoA. The enzyme is bound to adenosylcobalamin, a hormonal derivative of vitamin B12 in order to function. Methylmalonyl-CoA mutase deficiency is caused by genetic defect in the MUT gene responsible for encoding the enzyme. Deficiency in this enzyme accounts for 60% of the cases of methylmalonic acidemia.
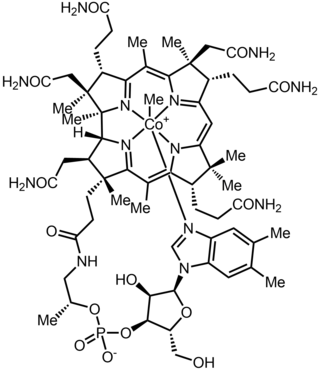
Methylcobalamin (mecobalamin, MeCbl, or MeB12) is a cobalamin, a form of vitamin B12. It differs from cyanocobalamin in that the cyano group at the cobalt is replaced with a methyl group. Methylcobalamin features an octahedral cobalt(III) centre and can be obtained as bright red crystals. From the perspective of coordination chemistry, methylcobalamin is notable as a rare example of a compound that contains metal–alkyl bonds. Nickel–methyl intermediates have been proposed for the final step of methanogenesis.
Methylmalonyl-CoA mutase (EC 5.4.99.2, MCM), mitochondrial, also known as methylmalonyl-CoA isomerase, is a protein that in humans is encoded by the MUT gene. This vitamin B12-dependent enzyme catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA in humans. Mutations in MUT gene may lead to various types of methylmalonic aciduria.

Methylmalonyl-CoA is the thioester consisting of coenzyme A linked to methylmalonic acid. It is an important intermediate in the biosynthesis of succinyl-CoA, which plays an essential role in the tricarboxylic acid cycle. The compound is sometimes referred to as "methylmalyl-CoA".

Adenosylcobalamin (AdoCbl), also known as coenzyme B12, cobamamide, and dibencozide, is, along with methylcobalamin (MeCbl), one of the biologically active forms of vitamin B12.
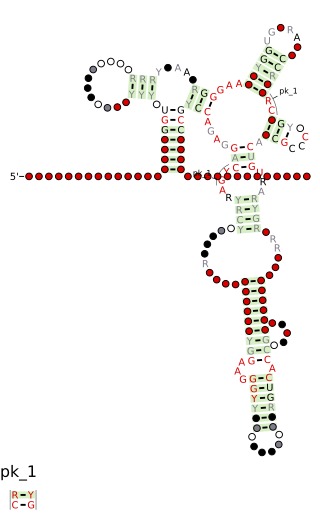
Cobalamin riboswitch is a cis-regulatory element which is widely distributed in 5' untranslated regions of vitamin B12 (Cobalamin) related genes in bacteria.

Cyanocobalamin is a form of vitamin B
12 used to treat and prevent vitamin B
12 deficiency except in the presence of cyanide toxicity. The deficiency may occur in pernicious anemia, following surgical removal of the stomach, with fish tapeworm, or due to bowel cancer. It is used by mouth, by injection into a muscle, or as a nasal spray.

Methionine synthase reductase, also known as MSR, is an enzyme that in humans is encoded by the MTRR gene.

D-2-hydroxyglutarate dehydrogenase, mitochondrial is an enzyme that in humans is encoded by the D2HGDH gene.

Dihydropyrimidinase is an enzyme that in humans is encoded by the DPYS gene.

Cob(I)yrinic acid a,c-diamide adenosyltransferase, mitochondrial is an enzyme that in humans is encoded by the MMAB gene.

Probable lysosomal cobalamin transporter is a protein that in humans is encoded by the LMBRD1 gene.

Methylmalonic aciduria type A protein, mitochondrial also known as MMAA is a protein that in humans is encoded by the MMAA gene.
Methylmalonic aciduria and homocystinuria type D protein, mitochondrial also known as MMADHC is a protein that in humans is encoded by the MMADHC gene.

Cubilin is a protein that in humans is encoded by the CUBN gene.
Cobalamin biosynthesis is the process by which bacteria and archea make cobalamin, vitamin B12. Many steps are involved in converting aminolevulinic acid via uroporphyrinogen III and adenosylcobyric acid to the final forms in which it is used by enzymes in both the producing organisms and other species, including humans who acquire it through their diet.
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid. However, the methylmalonic acid levels exceed those of malonic acid. Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism. Due to being infrequently diagnosed, it most often goes undetected.



