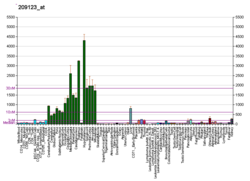
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.

Tetrahydrobiopterin (BH4, THB), also known as sapropterin (INN), is a cofactor of the three aromatic amino acid hydroxylase enzymes, used in the degradation of amino acid phenylalanine and in the biosynthesis of the neurotransmitters serotonin (5-hydroxytryptamine, 5-HT), melatonin, dopamine, norepinephrine (noradrenaline), epinephrine (adrenaline), and is a cofactor for the production of nitric oxide (NO) by the nitric oxide syntheses. Chemically, its structure is that of a (dihydropteridine reductase) reduced pteridine derivative (Quinonoid dihydrobiopterin).

Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.
Tyrosine hydroxylase or tyrosine 3-monooxygenase is the enzyme responsible for catalyzing the conversion of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA). It does so using molecular oxygen (O2), as well as iron (Fe2+) and tetrahydrobiopterin as cofactors. L-DOPA is a precursor for dopamine, which, in turn, is a precursor for the important neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline). Tyrosine hydroxylase catalyzes the rate limiting step in this synthesis of catecholamines. In humans, tyrosine hydroxylase is encoded by the TH gene, and the enzyme is present in the central nervous system (CNS), peripheral sympathetic neurons and the adrenal medulla. Tyrosine hydroxylase, phenylalanine hydroxylase and tryptophan hydroxylase together make up the family of aromatic amino acid hydroxylases (AAAHs).

Arylsulfatase A is an enzyme that breaks down sulfatides, namely cerebroside 3-sulfate into cerebroside and sulfate. In humans, arylsulfatase A is encoded by the ARSA gene.

Steroid 11β-hydroxylase, also known as steroid 11β-monooxygenase, is a steroid hydroxylase found in the zona glomerulosa and zona fasciculata of the adrenal cortex. Named officially the cytochrome P450 11B1, mitochondrial, it is a protein that in humans is encoded by the CYP11B1 gene. The enzyme is involved in the biosynthesis of adrenal corticosteroids by catalyzing the addition of hydroxyl groups during oxidation reactions.

N-acetylgalactosamine-6-sulfatase is an enzyme that, in humans, is encoded by the GALNS gene.

Cartilage associated protein is a protein that in humans is encoded by the CRTAP gene.

Sepiapterin reductase is an enzyme that in humans is encoded by the SPR gene.
In enzymology, 6,7-dihydropteridine reductase (EC 1.5.1.34, also Dihydrobiopterin reductase) is an enzyme that catalyzes the chemical reaction

6-pyruvoyltetrahydropterin synthase, also known as PTS, is a human gene which facilitates folate biosynthesis.

Serine—pyruvate aminotransferase is an enzyme that in humans is encoded by the AGXT gene.

Phosphorylase b kinase regulatory subunit alpha, liver isoform is an enzyme that in humans is encoded by the PHKA2 gene.

GTP cyclohydrolase 1 feedback regulatory protein is an enzyme that in humans is encoded by the GCHFR gene.

3-keto-steroid reductase is an enzyme that in humans is encoded by the HSD17B7 gene.
Phosphorylase b kinase regulatory subunit beta is an enzyme that in humans is encoded by the PHKB gene.

Glyoxylate reductase/hydroxypyruvate reductase is an enzyme that in humans is encoded by the GRHPR gene.

Para-hydroxybenzoate—polyprenyltransferase, mitochondrial is an enzyme that in humans is encoded by the COQ2 gene.

CYP2R1 is cytochrome P450 2R1, an enzyme which is the principal vitamin D 25-hydroxylase. In humans it is encoded by the CYP2R1 gene located on chromosome 11p15.2. It is expressed in the endoplasmic reticulum in liver, where it performs the first step in the activation of vitamin D by catalyzing the formation of 25-hydroxyvitamin D.
Dihydropteridine reductase deficiency (DHPRD) is a genetic disorder affecting the tetrahydrobiopterin (BH4) synthesis pathway, inherited in the autosomal recessive pattern. It is one of the six known disorders causing tetrahydrobiopterin deficiency, and occurs in patients with mutations of the QDPR gene.