
Mastocytosis, a type of mast cell disease, is a rare disorder affecting both children and adults caused by the accumulation of functionally defective mast cells and CD34+ mast cell precursors.

Liposarcomas are the most common subtype of soft tissue sarcomas, accounting for at least 20% of all sarcomas in adults. Soft tissue sarcomas are rare neoplasms with over 150 different histological subtypes or forms. Liposarcomas arise from the precursor lipoblasts of the adipocytes in adipose tissues. Adipose tissues are distributed throughout the body, including such sites as the deep and more superficial layers of subcutaneous tissues as well as in less surgically accessible sites like the retroperitoneum and visceral fat inside the abdominal cavity.

Nodular fasciitis (NF) is a benign, soft tissue tumor composed of myofibroblasts that typically occurs in subcutaneous tissue, fascia, and/or muscles. The literature sometimes titles rare NF variants according to their tissue locations. The most frequently used and important of these are cranial fasciitis and intravascular fasciitis. In 2020, the World Health Organization classified nodular fasciitis as in the category of benign fibroblastic/myofibroblastic tumors. NF is the most common of the benign fibroblastic proliferative tumors of soft tissue.
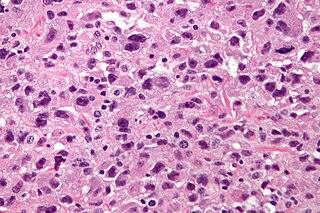
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.
The Ewing family of tumors (EFTs) is a group of small cell sarcomas including Ewing sarcoma of the bone, extra osseous Ewing tumors, and primitive neuroectodermal tumors. They are rare cancers, usually diagnosed in peoples' twenties. The sarcoma of bone is the most common of the variants. All forms are predisposed to metastasis and have had historically high rates of mortality. The family of tumors shares a common translocation mutation of the EWS gene on chromosome 22 to an ETS-type gene, most commonly the FLI1 gene. EFTs are highly malignant, with 5-year survival for patients with metastatic disease at 20%. The current standard of care includes resection, radiation, and chemotherapy.

Mast cell leukemia is an extremely aggressive subtype of acute myeloid leukemia that usually occurs de novo but can, rarely, evolve from transformation of chronic myeloid leukemia into the more aggressive acute myeloid leukemia. In a small proportion of cases, acute mast cell leukemia may evolve from a more progressive form of systemic mastocytosis. The diagnosis of acute mast cell leukemia by the WHO criteria includes the requirement for a prevalence of 20% neoplastic mast cells in marrow and 10% in blood. If the mast cells represent less than 10% of blood cells, the tumor is called "aleukemic" mast cell leukemia.
Fibrous hamartoma of infancy (FHI) is a rare, typically painless, benign tumor that develops in the subcutaneous tissues of the axilla, arms, external genitalia, or, less commonly, various other areas. It is diagnosed in children who are usually less than 2 years old or, in up to 20% of cases, develops in utero and is diagnosed in an infant at birth.

Follicular dendritic cell sarcoma (FDCS) is an extremely rare neoplasm. While the existence of FDC tumors was predicted by Lennert in 1978, the tumor wasn't fully recognized as its own cancer until 1986 after characterization by Monda et al. It accounts for only 0.4% of soft tissue sarcomas, but has significant recurrent and metastatic potential and is considered an intermediate grade malignancy. The major hurdle in treating FDCS has been misdiagnosis. It is a newly characterized cancer, and because of its similarities in presentation and markers to lymphoma, both Hodgkin and Non-Hodgkin subtypes, diagnosis of FDCS can be difficult. With recent advancements in cancer biology better diagnostic assays and chemotherapeutic agents have been made to more accurately diagnose and treat FDCS.
Mucinous cystadenocarcinoma of the lung (MCACL) is a very rare malignant mucus-producing neoplasm arising from the uncontrolled growth of transformed epithelial cells originating in lung tissue.

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.

Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of the mesodermal cells that form the connective tissues which support virtually all of the organs and tissues of the body. IMT was formerly termed inflammatory pseudotumor. Currently, however, inflammatory pseudotumor designates a large and heterogeneous group of soft tissue tumors that includes inflammatory myofibroblastic tumor, plasma cell granuloma, xanthomatous pseudotumor, solitary mast cell granuloma, inflammatory fibrosarcoma, pseudosarcomatous myofibroblastic proliferation, myofibroblastoma, inflammatory myofibrohistiocytic proliferation, and other tumors that develop from connective tissue cells. Inflammatory pseudotumour is a generic term applied to various neoplastic and non-neoplastic tissue lesions which share a common microscopic appearance consisting of spindle cells and a prominent presence of the white blood cells that populate chronic or, less commonly, acute inflamed tissues.

Mammary-type myofibroblastoma (MFB), also named mammary and extramammary myofibroblastoma, was first termed myofibrolastoma of the breast, or, more simply, either mammary myofibroblastoma (MMFB) or just myofibroblastoma. The change in this terminology occurred because the initial 1987 study and many subsequent studies found this tumor only in breast tissue. However, a 2001 study followed by numerous reports found tumors with the microscopic histopathology and other key features of mammary MFB in a wide range of organs and tissues. Further complicating the issue, early studies on MFB classified it as one of various types of spindle cell tumors that, except for MFB, were ill-defined. These other tumors, which have often been named interchangeably in different reports, are: myelofibroblastoma, benign spindle cell tumor, fibroma, spindle cell lipoma, myogenic stromal tumor, and solitary stromal tumor. Finally, studies suggest that spindle cell lipoma and cellular angiofibroma are variants of MFB. Here, the latter two tumors are tentatively classified as MFB variants but otherwise MFB is described as it is more strictly defined in most recent publications. The World Health Organization in 2020 classified mammary type myofibroblastoma tumors and myofibroblastoma tumors as separate tumor forms within the category of fibroblastic and myofibroblastic tumors.
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
Acral myxoinflammatory fibroblastic sarcoma (AMSF), also termed myxoinflammatory fibroblastic sarcoma (MSF), is a rare, low-grade, soft tissue tumor that the World Health Organization (2020) classified as in the category of rarely metastasizing fibroblastic and myofibroblastic tumors. It is a locally aggressive neoplasm that often recurs at the site of its surgical removal. However, it usually grows slowly and in only 1–2% of cases spreads to distant tissues.
Peter Valent is an Austrian hematologist and stem cell researcher. Since 1990 he leads a research group at the Medical University of Vienna. From 2002 he coordinates the European Competence Network on Mastocytosis and since 2008 he is Scientific Director of the Ludwig Boltzmann Institute for Hematology and Oncology of the Ludwig Boltzmann Society in Austria.
Fibroblastic and myofibroblastic tumors (FMTs) develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
Sclerosing epithelioid fibrosarcoma (SEF) is a very rare malignant tumor of soft tissues that on microscopic examination consists of small round or ovoid neoplastic epithelioid fibroblast-like cells, i.e. cells that have features resembling both epithelioid cells and fibroblasts. In 2020, the World Health Organization classified SEF as a distinct tumor type in the category of malignant fibroblastic and myofibroblastic tumors. However, current studies have reported that low-grade fibromyxoid sarcoma (LGFMS) has many clinically and pathologically important features characteristic of SEF; these studies suggest that LGSFMS may be an early form of, and over time progress to become, a SEF. Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF, SEF and LGFMS are here regarded as different tumor forms.
The FET protein family consists of three similarly structured and functioning proteins. They and the genes in the FET gene family which encode them are: 1) the EWSR1 protein encoded by the EWSR1 gene located at band 12.2 of the long arm of chromosome 22; 2) the FUS protein encoded by the FUS gene located at band 16 on the short arm of chromosome 16; and 3) the TAF15 protein encoded by the TAF15 gene located at band 12 on the long arm of chromosome 7 The FET in this protein family's name derives from the first letters of FUS, EWSR1, and TAF15.
Cellular angiofibroma (CAF) is a rare, benign tumor of superficial soft tissues that was first described by M. R. Nucci et al. in 1997. These tumors occur predominantly in the distal parts of the female and male reproductive systems, i.e. in the vulva-vaginal and inguinal-scrotal areas, respectively, or, less commonly, in various other superficial soft tissue areas throughout the body. CAF tumors develop exclusively in adults who typically are more than 30 years old.
