A coordination complex consists of a central atom or ion, which is usually metallic and is called the coordination centre, and a surrounding array of bound molecules or ions, that are in turn known as ligands or complexing agents. Many metal-containing compounds, especially those that include transition metals, are coordination complexes.

Auger electron spectroscopy is a common analytical technique used specifically in the study of surfaces and, more generally, in the area of materials science. It is a form of electron spectroscopy that relies on the Auger effect, based on the analysis of energetic electrons emitted from an excited atom after a series of internal relaxation events. The Auger effect was discovered independently by both Lise Meitner and Pierre Auger in the 1920s. Though the discovery was made by Meitner and initially reported in the journal Zeitschrift für Physik in 1922, Auger is credited with the discovery in most of the scientific community. Until the early 1950s Auger transitions were considered nuisance effects by spectroscopists, not containing much relevant material information, but studied so as to explain anomalies in X-ray spectroscopy data. Since 1953 however, AES has become a practical and straightforward characterization technique for probing chemical and compositional surface environments and has found applications in metallurgy, gas-phase chemistry, and throughout the microelectronics industry.

Magnetic circular dichroism (MCD) is the differential absorption of left and right circularly polarized light, induced in a sample by a strong magnetic field oriented parallel to the direction of light propagation. MCD measurements can detect transitions which are too weak to be seen in conventional optical absorption spectra, and it can be used to distinguish between overlapping transitions. Paramagnetic systems are common analytes, as their near-degenerate magnetic sublevels provide strong MCD intensity that varies with both field strength and sample temperature. The MCD signal also provides insight into the symmetry of the electronic levels of the studied systems, such as metal ion sites.
Crystal field theory (CFT) describes the breaking of degeneracies of electron orbital states, usually d or f orbitals, due to a static electric field produced by a surrounding charge distribution. This theory has been used to describe various spectroscopies of transition metal coordination complexes, in particular optical spectra (colors). CFT successfully accounts for some magnetic properties, colors, hydration enthalpies, and spinel structures of transition metal complexes, but it does not attempt to describe bonding. CFT was developed by physicists Hans Bethe and John Hasbrouck van Vleck in the 1930s. CFT was subsequently combined with molecular orbital theory to form the more realistic and complex ligand field theory (LFT), which delivers insight into the process of chemical bonding in transition metal complexes.
Ligand field theory (LFT) describes the bonding, orbital arrangement, and other characteristics of coordination complexes. It represents an application of molecular orbital theory to transition metal complexes. A transition metal ion has nine valence atomic orbitals - consisting of five nd, one (n+1)s, and three (n+1)p orbitals. These orbitals are of appropriate energy to form bonding interaction with ligands. The LFT analysis is highly dependent on the geometry of the complex, but most explanations begin by describing octahedral complexes, where six ligands coordinate to the metal. Other complexes can be described by reference to crystal field theory.

In chemistry, π backbonding, also called π backdonation, is when electrons move from an atomic orbital on one atom to an appropriate symmetry antibonding orbital on a π-acceptor ligand. It is especially common in the organometallic chemistry of transition metals with multi-atomic ligands such as carbon monoxide, ethylene or the nitrosonium cation. Electrons from the metal are used to bond to the ligand, in the process relieving the metal of excess negative charge. Compounds where π backbonding occurs include Ni(CO)4 and Zeise's salt. IUPAC offers the following definition for backbonding:
A description of the bonding of π-conjugated ligands to a transition metal which involves a synergic process with donation of electrons from the filled π-orbital or lone electron pair orbital of the ligand into an empty orbital of the metal (donor–acceptor bond), together with release (back donation) of electrons from an nd orbital of the metal (which is of π-symmetry with respect to the metal–ligand axis) into the empty π*-antibonding orbital of the ligand.

Metal carbonyls are coordination complexes of transition metals with carbon monoxide ligands. Metal carbonyls are useful in organic synthesis and as catalysts or catalyst precursors in homogeneous catalysis, such as hydroformylation and Reppe chemistry. In the Mond process, nickel tetracarbonyl is used to produce pure nickel. In organometallic chemistry, metal carbonyls serve as precursors for the preparation of other organometallic complexes.
A spectrochemical series is a list of ligands ordered by ligand "strength", and a list of metal ions based on oxidation number, group and element. For a metal ion, the ligands modify the difference in energy Δ between the d orbitals, called the ligand-field splitting parameter in ligand field theory, or the crystal-field splitting parameter in crystal field theory. The splitting parameter is reflected in the ion's electronic and magnetic properties such as its spin state, and optical properties such as its color and absorption spectrum.
Copper proteins are proteins that contain one or more copper ions as prosthetic groups. Copper proteins are found in all forms of air-breathing life. These proteins are usually associated with electron-transfer with or without the involvement of oxygen (O2). Some organisms even use copper proteins to carry oxygen instead of iron proteins. A prominent copper proteins in humans is in cytochrome c oxidase (cco). The enzyme cco mediates the controlled combustion that produces ATP.
X-ray absorption near edge structure (XANES), also known as near edge X-ray absorption fine structure (NEXAFS), is a type of absorption spectroscopy that indicates the features in the X-ray absorption spectra (XAS) of condensed matter due to the photoabsorption cross section for electronic transitions from an atomic core level to final states in the energy region of 50–100 eV above the selected atomic core level ionization energy, where the wavelength of the photoelectron is larger than the interatomic distance between the absorbing atom and its first neighbour atoms.
In coordination chemistry, Tanabe–Sugano diagrams are used to predict absorptions in the ultraviolet (UV), visible and infrared (IR) electromagnetic spectrum of coordination compounds. The results from a Tanabe–Sugano diagram analysis of a metal complex can also be compared to experimental spectroscopic data. They are qualitatively useful and can be used to approximate the value of 10Dq, the ligand field splitting energy. Tanabe–Sugano diagrams can be used for both high spin and low spin complexes, unlike Orgel diagrams, which apply only to high spin complexes. Tanabe–Sugano diagrams can also be used to predict the size of the ligand field necessary to cause high-spin to low-spin transitions.

Mössbauer spectroscopy is a spectroscopic technique based on the Mössbauer effect. This effect, discovered by Rudolf Mössbauer in 1958, consists of the nearly recoil-free emission and absorption of nuclear gamma rays in solids. The consequent nuclear spectroscopy method is exquisitely sensitive to small changes in the chemical environment of certain nuclei.
In X-ray absorption spectroscopy, the K-edge is a sudden increase in x-ray absorption occurring when the energy of the X-rays is just above the binding energy of the innermost electron shell of the atoms interacting with the photons. The term is based on X-ray notation, where the innermost electron shell is known as the K-shell. Physically, this sudden increase in attenuation is caused by the photoelectric absorption of the photons. For this interaction to occur, the photons must have more energy than the binding energy of the K-shell electrons (K-edge). A photon having an energy just above the binding energy of the electron is therefore more likely to be absorbed than a photon having an energy just below this binding energy or significantly above it.
Spin states when describing transition metal coordination complexes refers to the potential spin configurations of the central metal's d electrons. For several oxidation states, metals can adopt high-spin and low-spin configurations. The ambiguity only applies to first row metals, because second- and third-row metals are invariably low-spin. These configurations can be understood through the two major models used to describe coordination complexes; crystal field theory and ligand field theory.
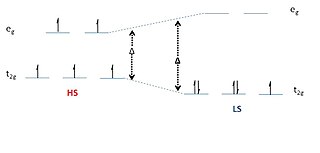
Spin crossover (SCO) is a phenomenon that occurs in some metal complexes wherein the spin state of the complex changes due to an external stimulus. The stimuli can include temperature or pressure. Spin crossover is sometimes referred to as spin transition or spin equilibrium behavior. The change in spin state usually involves interchange of low spin (LS) and high spin (HS) configuration.

Resonant inelastic X-ray scattering (RIXS) is an X-ray spectroscopy technique used to investigate the electronic structure of molecules and materials.

The Tolman electronic parameter (TEP) is a measure of the electron donating or withdrawing ability of a ligand. It is determined by measuring the frequency of the A1 C-O vibrational mode (ν(CO)) of a (pseudo)-C3v symmetric complex, [LNi(CO)3] by infrared spectroscopy, where L is the ligand of interest. [LNi(CO)3] was chosen as the model compound because such complexes are readily prepared from tetracarbonylnickel(0). The shift in ν(CO) is used to infer the electronic properties of a ligand, which can aid in understanding its behavior in other complexes. The analysis was introduced by Chadwick A. Tolman.

Charge-transfer bands are a characteristic feature of the optical spectra of many compounds. These bands are typically more intense than d–d transitions. They typically exhibit solvatochromism, consistent with shifts of electron density that would be sensitive to solvation.

X-ray emission spectroscopy (XES) is a form of X-ray spectroscopy in which the X-ray line spectra are measured with a spectral resolution sufficient to analyze the impact of the chemical environment on the X-ray line energy and on branching ratios. This is done by exciting electrons out of their shell and then watching the emitted photons of the recombinating electrons.

Alkaline earth octacarbonyl complexes are a class of neutral compounds that have the general formula M(CO)8 where M is a heavy Group 2 element (Ca, Sr, or Ba). The metal center has a formal oxidation state of 0 and the complex has a high level of symmetry belonging to the cubic Oh point group. These complexes are isolable in a low-temperature neon matrix, but are not frequently used in applications due to their instability in air and water. The bonding within these complexes is controversial with some arguing the bonding resembles a model similar to bonding in transition metal carbonyl complexes which abide by the 18-electron rule, and others arguing the molecule more accurately contains ionic bonds between the alkaline earth metal center and the carbonyl ligands. Complexes of Be(CO)8 and Mg(CO)8 are not synthetically possible due to inaccessible (n-1)d orbitals. Beryllium has been found to form a dinuclear homoleptic carbonyl and magnesium a mononuclear heteroleptic carbonyl, both with only two carbonyl ligands instead of eight to each metal atom.
![Figure 1: L3- and L2-edges of [CuCl4] . CuCl4-Ledge.svg](http://upload.wikimedia.org/wikipedia/en/thumb/d/d8/CuCl4-Ledge.svg/220px-CuCl4-Ledge.svg.png)


![Figure 4: Comparison of the Fe L-edges of low-spin K3[Fe(CN)6] and [Fe(tacn)2]Cl3. Tacn is a s-only donor, meaning no backbonding and only two main L-edge features. K3[Fe(CN)6] has significant backbonding, as is shown by the third transition to higher energy in the L-edge spectrum. Ferric L-edge.png](http://upload.wikimedia.org/wikipedia/en/thumb/0/0b/Ferric_L-edge.png/220px-Ferric_L-edge.png)