
Diatomic molecules are molecules composed of only two atoms, of the same or different chemical elements. If a diatomic molecule consists of two atoms of the same element, such as hydrogen or oxygen, then it is said to be homonuclear. Otherwise, if a diatomic molecule consists of two different atoms, such as carbon monoxide or nitric oxide, the molecule is said to be heteronuclear. The bond in a homonuclear diatomic molecule is non-polar.

In physics, a dipole is an electromagnetic phenomenon which occurs in two ways:
Circular dichroism (CD) is dichroism involving circularly polarized light, i.e., the differential absorption of left- and right-handed light. Left-hand circular (LHC) and right-hand circular (RHC) polarized light represent two possible spin angular momentum states for a photon, and so circular dichroism is also referred to as dichroism for spin angular momentum. This phenomenon was discovered by Jean-Baptiste Biot, Augustin Fresnel, and Aimé Cotton in the first half of the 19th century. Circular dichroism and circular birefringence are manifestations of optical activity. It is exhibited in the absorption bands of optically active chiral molecules. CD spectroscopy has a wide range of applications in many different fields. Most notably, UV CD is used to investigate the secondary structure of proteins. UV/Vis CD is used to investigate charge-transfer transitions. Near-infrared CD is used to investigate geometric and electronic structure by probing metal d→d transitions. Vibrational circular dichroism, which uses light from the infrared energy region, is used for structural studies of small organic molecules, and most recently proteins and DNA.
Rotational–vibrational spectroscopy is a branch of molecular spectroscopy that is concerned with infrared and Raman spectra of molecules in the gas phase. Transitions involving changes in both vibrational and rotational states can be abbreviated as rovibrational transitions. When such transitions emit or absorb photons, the frequency is proportional to the difference in energy levels and can be detected by certain kinds of spectroscopy. Since changes in rotational energy levels are typically much smaller than changes in vibrational energy levels, changes in rotational state are said to give fine structure to the vibrational spectrum. For a given vibrational transition, the same theoretical treatment as for pure rotational spectroscopy gives the rotational quantum numbers, energy levels, and selection rules. In linear and spherical top molecules, rotational lines are found as simple progressions at both higher and lower frequencies relative to the pure vibration frequency. In symmetric top molecules the transitions are classified as parallel when the dipole moment change is parallel to the principal axis of rotation, and perpendicular when the change is perpendicular to that axis. The ro-vibrational spectrum of the asymmetric rotor water is important because of the presence of water vapor in the atmosphere.

In atomic physics, hyperfine structure is defined by small shifts in otherwise degenerate energy levels and the resulting splittings in those energy levels of atoms, molecules, and ions, due to electromagnetic multipole interaction between the nucleus and electron clouds.

Rotational spectroscopy is concerned with the measurement of the energies of transitions between quantized rotational states of molecules in the gas phase. The rotational spectrum of polar molecules can be measured in absorption or emission by microwave spectroscopy or by far infrared spectroscopy. The rotational spectra of non-polar molecules cannot be observed by those methods, but can be observed and measured by Raman spectroscopy. Rotational spectroscopy is sometimes referred to as pure rotational spectroscopy to distinguish it from rotational-vibrational spectroscopy where changes in rotational energy occur together with changes in vibrational energy, and also from ro-vibronic spectroscopy where rotational, vibrational and electronic energy changes occur simultaneously.

In electromagnetism, the magnetic moment or magnetic dipole moment is the combination of strength and orientation of a magnet or other object or system that exerts a magnetic field. The magnetic dipole moment of an object determines the magnitude of torque the object experiences in a given magnetic field. When the same magnetic field is applied, objects with larger magnetic moments experience larger torques. The strength of this torque depends not only on the magnitude of the magnetic moment but also on its orientation relative to the direction of the magnetic field. Its direction points from the south pole to north pole of the magnet.
In atomic physics, a magnetic quantum number is a quantum number used to distinguish quantum states of an electron or other particle according to its angular momentum along a given axis in space. The orbital magnetic quantum number distinguishes the orbitals available within a given subshell of an atom. It specifies the component of the orbital angular momentum that lies along a given axis, conventionally called the z-axis, so it describes the orientation of the orbital in space. The spin magnetic quantum numberms specifies the z-axis component of the spin angular momentum for a particle having spin quantum number s. For an electron, s is 1⁄2, and ms is either +1⁄2 or −1⁄2, often called "spin-up" and "spin-down", or α and β. The term magnetic in the name refers to the magnetic dipole moment associated with each type of angular momentum, so states having different magnetic quantum numbers shift in energy in a magnetic field according to the Zeeman effect.
In physics, Raman scattering or the Raman effect is the inelastic scattering of photons by matter, meaning that there is both an exchange of energy and a change in the light's direction. Typically this effect involves vibrational energy being gained by a molecule as incident photons from a visible laser are shifted to lower energy. This is called normal Stokes-Raman scattering.
In quantum mechanics, a rotational transition is an abrupt change in angular momentum. Like all other properties of a quantum particle, angular momentum is quantized, meaning it can only equal certain discrete values, which correspond to different rotational energy states. When a particle loses angular momentum, it is said to have transitioned to a lower rotational energy state. Likewise, when a particle gains angular momentum, a positive rotational transition is said to have occurred.
In rotordynamics, the rigid rotor is a mechanical model of rotating systems. An arbitrary rigid rotor is a 3-dimensional rigid object, such as a top. To orient such an object in space requires three angles, known as Euler angles. A special rigid rotor is the linear rotor requiring only two angles to describe, for example of a diatomic molecule. More general molecules are 3-dimensional, such as water, ammonia, or methane.
A multipole expansion is a mathematical series representing a function that depends on angles—usually the two angles used in the spherical coordinate system for three-dimensional Euclidean space, . Similarly to Taylor series, multipole expansions are useful because oftentimes only the first few terms are needed to provide a good approximation of the original function. The function being expanded may be real- or complex-valued and is defined either on , or less often on for some other .
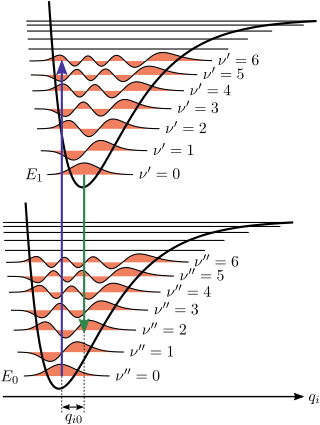
The Franck–Condon principle is a rule in spectroscopy and quantum chemistry that explains the intensity of vibronic transitions. The principle states that during an electronic transition, a change from one vibrational energy level to another will be more likely to happen if the two vibrational wave functions overlap more significantly.
In molecular physics, the molecular term symbol is a shorthand expression of the group representation and angular momenta that characterize the state of a molecule, i.e. its electronic quantum state which is an eigenstate of the electronic molecular Hamiltonian. It is the equivalent of the term symbol for the atomic case. However, the following presentation is restricted to the case of homonuclear diatomic molecules, or other symmetric molecules with an inversion centre. For heteronuclear diatomic molecules, the u/g symbol does not correspond to any exact symmetry of the electronic molecular Hamiltonian. In the case of less symmetric molecules the molecular term symbol contains the symbol of the group representation to which the molecular electronic state belongs.
High resolution electron energy loss spectroscopy (HREELS) is a tool used in surface science. The inelastic scattering of electrons from surfaces is utilized to study electronic excitations or vibrational modes of the surface of a material or of molecules adsorbed to a surface. In contrast to other electron energy loss spectroscopies (EELS), HREELS deals with small energy losses in the range of 10−3 eV to 1 eV. It plays an important role in the investigation of surface structure, catalysis, dispersion of surface phonons and the monitoring of epitaxial growth.
In spectroscopy, a forbidden mechanism is a spectral line associated with absorption or emission of photons by atomic nuclei, atoms, or molecules which undergo a transition that is not allowed by a particular selection rule but is allowed if the approximation associated with that rule is not made. For example, in a situation where, according to usual approximations, the process cannot happen, but at a higher level of approximation the process is allowed but at a low rate.
A molecular vibration is a periodic motion of the atoms of a molecule relative to each other, such that the center of mass of the molecule remains unchanged. The typical vibrational frequencies range from less than 1013 Hz to approximately 1014 Hz, corresponding to wavenumbers of approximately 300 to 3000 cm−1 and wavelengths of approximately 30 to 3 µm.
In rotational-vibrational and electronic spectroscopy of diatomic molecules, Hund's coupling cases are idealized descriptions of rotational states in which specific terms in the molecular Hamiltonian and involving couplings between angular momenta are assumed to dominate over all other terms. There are five cases, proposed by Friedrich Hund in 1926-27 and traditionally denoted by the letters (a) through (e). Most diatomic molecules are somewhere between the idealized cases (a) and (b).
Molecular symmetry in physics and chemistry describes the symmetry present in molecules and the classification of molecules according to their symmetry. Molecular symmetry is a fundamental concept in the application of Quantum Mechanics in physics and chemistry, for example it can be used to predict or explain many of a molecule's properties, such as its dipole moment and its allowed spectroscopic transitions, without doing the exact rigorous calculations. To do this it is necessary to classify the states of the molecule using the irreducible representations from the character table of the symmetry group of the molecule. Among all the molecular symmetries, diatomic molecules show some distinct features and they are relatively easier to analyze.
Stimulated Raman spectroscopy, also referred to as stimulated Raman scattering (SRS) is a form of spectroscopy employed in physics, chemistry, biology, and other fields. The basic mechanism resembles that of spontaneous Raman spectroscopy: a pump photon, of the angular frequency , which is scattered by a molecule has some small probability of inducing some vibrational transition, as opposed to inducing a simple Rayleigh transition. This makes the molecule emit a photon at a shifted frequency. However, SRS, as opposed to spontaneous Raman spectroscopy, is a third-order non-linear phenomenon involving a second photon—the Stokes photon of angular frequency —which stimulates a specific transition. When the difference in frequency between both photons resembles that of a specific vibrational transition the occurrence of this transition is resonantly enhanced. In SRS, the signal is equivalent to changes in the intensity of the pump and Stokes beams. The signals are typically rather low, of the order of a part in 10^5, thus calling for modulation-transfer techniques: one beam is modulated in amplitude and the signal is detected on the other beam via a lock-in amplifier. Employing a pump laser beam of a constant frequency and a Stokes laser beam of a scanned frequency allows for the unraveling of the spectral fingerprint of the molecule. This spectral fingerprint differs from those obtained by other spectroscopy methods such as Rayleigh scattering as the Raman transitions confer to different exclusion rules than those that apply for Rayleigh transitions.

















































