Brunauer, Emmett and Teller's model of multilayer adsorption is a random distribution of molecules on the material surface.
Adsorption is the adhesion[1] of atoms, ions or molecules from a gas, liquid or dissolved solid to a surface.[2] This process creates a film of the adsorbate on the surface of the adsorbent. This process differs from absorption, in which a fluid (the absorbate) is dissolved by or permeates a liquid or solid (the absorbent).[3] While adsorption does often precede absorption, which involves the transfer of the absorbate into the volume of the absorbent material, alternatively, adsorption is distinctly a surface phenomenon, wherein the adsorbate does not penetrate through the material surface and into the bulk of the adsorbent.[4] The term sorption encompasses both adsorption and absorption, and desorption is the reverse of sorption.
adsorption: An increase in the concentration of a dissolved substance at the interface of a condensed and a liquid phase due to the operation of surface forces. Adsorption can also occur at the interface of a condensed and a gaseous phase. [5]
Like surface tension, adsorption is a consequence of surface energy. In a bulk material, all the bonding requirements (be they ionic, covalent or metallic) of the constituent atoms of the material are fulfilled by other atoms in the material. However, atoms on the surface of the adsorbent are not wholly surrounded by other adsorbent atoms and therefore can attract adsorbates. The exact nature of the bonding depends on the details of the species involved, but the adsorption process is generally classified as physisorption (characteristic of weak van der Waals forces) or chemisorption (characteristic of covalent bonding). It may also occur due to electrostatic attraction.[6][7] The nature of the adsorption can affect the structure of the adsorbed species. For example, polymer physisorption from solution can result in squashed structures on a surface.[8]
Adsorption is present in many natural, physical, biological and chemical systems and is widely used in industrial applications such as heterogeneous catalysts,[9][10]activated charcoal, capturing and using waste heat to provide cold water for air conditioning and other process requirements (adsorption chillers), synthetic resins, increasing storage capacity of carbide-derived carbons and water purification. Adsorption, ion exchange and chromatography are sorption processes in which certain adsorbates are selectively transferred from the fluid phase to the surface of insoluble, rigid particles suspended in a vessel or packed in a column. Pharmaceutical industry applications, which use adsorption as a means to prolong neurological exposure to specific drugs or parts thereof,[citation needed] are lesser known.
The word "adsorption" was coined in 1881 by German physicist Heinrich Kayser (1853–1940).[11]
Isotherms
The adsorption of gases and solutes is usually described through isotherms, that is, the amount of adsorbate on the adsorbent as a function of its pressure (if gas) or concentration (for liquid phase solutes) at constant temperature. The quantity adsorbed is nearly always normalized by the mass of the adsorbent to allow comparison of different materials. To date, 15 different isotherm models have been developed.[12]
The first mathematical fit to an isotherm was published by Freundlich and Kuster (1906) and is a purely empirical formula for gaseous adsorbates:
where is the mass of adsorbate adsorbed, is the mass of the adsorbent, is the pressure of adsorbate (this can be changed to concentration if investigating solution rather than gas), and and are empirical constants for each adsorbent–adsorbate pair at a given temperature. The function is not adequate at very high pressure because in reality has an asymptotic maximum as pressure increases without bound. As the temperature increases, the constants and change to reflect the empirical observation that the quantity adsorbed rises more slowly and higher pressures are required to saturate the surface.
Irving Langmuir was the first to derive a scientifically based adsorption isotherm in 1918.[13] The model applies to gases adsorbed on solid surfaces. It is a semi-empirical isotherm with a kinetic basis and was derived based on statistical thermodynamics. It is the most common isotherm equation to use due to its simplicity and its ability to fit a variety of adsorption data. It is based on four assumptions:
All of the adsorption sites are equivalent, and each site can only accommodate one molecule.
The surface is energetically homogeneous, and adsorbed molecules do not interact.
At the maximum adsorption, only a monolayer is formed. Adsorption only occurs on localized sites on the surface, not with other adsorbates.
These four assumptions are seldom all true: there are always imperfections on the surface, adsorbed molecules are not necessarily inert, and the mechanism is clearly not the same for the first molecules to adsorb to a surface as for the last. The fourth condition is the most troublesome, as frequently more molecules will adsorb to the monolayer; this problem is addressed by the BET isotherm for relatively flat (non-microporous) surfaces. The Langmuir isotherm is nonetheless the first choice for most models of adsorption and has many applications in surface kinetics (usually called Langmuir–Hinshelwood kinetics) and thermodynamics.
Langmuir suggested that adsorption takes place through this mechanism: , where A is a gas molecule, and S is an adsorption site. The direct and inverse rate constants are k and k−1. If we define surface coverage, , as the fraction of the adsorption sites occupied, in the equilibrium we have:
or
where is the partial pressure of the gas or the molar concentration of the solution. For very low pressures , and for high pressures .
The value of is difficult to measure experimentally; usually, the adsorbate is a gas and the quantity adsorbed is given in moles, grams, or gas volumes at standard temperature and pressure (STP) per gram of adsorbent. If we call vmon the STP volume of adsorbate required to form a monolayer on the adsorbent (per gram of adsorbent), then , and we obtain an expression for a straight line:
Through its slope and y intercept we can obtain vmon and K, which are constants for each adsorbent–adsorbate pair at a given temperature. vmon is related to the number of adsorption sites through the ideal gas law. If we assume that the number of sites is just the whole area of the solid divided into the cross section of the adsorbate molecules, we can easily calculate the surface area of the adsorbent. The surface area of an adsorbent depends on its structure: the more pores it has, the greater the area, which has a big influence on reactions on surfaces.
If more than one gas adsorbs on the surface, we define as the fraction of empty sites, and we have:
Also, we can define as the fraction of the sites occupied by the j-th gas:
where i is each one of the gases that adsorb.
Note:
1) To choose between the Langmuir and Freundlich equations, the enthalpies of adsorption must be investigated.[14] While the Langmuir model assumes that the energy of adsorption remains constant with surface occupancy, the Freundlich equation is derived with the assumption that the heat of adsorption continually decrease as the binding sites are occupied.[15] The choice of the model based on best fitting of the data is a common misconception.[14]
2) The use of the linearized form of the Langmuir model is no longer common practice. Advances in computational power allowed for nonlinear regression to be performed quickly and with higher confidence since no data transformation is required.
Often molecules do form multilayers, that is, some are adsorbed on already adsorbed molecules, and the Langmuir isotherm is not valid. In 1938 Stephen Brunauer, Paul Emmett, and Edward Teller developed a model isotherm that takes that possibility into account. Their theory is called BET theory, after the initials in their last names. They modified Langmuir's mechanism as follows:
A(g) + S ⇌ AS,
A(g) + AS ⇌ A2S,
A(g) + A2S ⇌ A3S and so on.
Langmuir (blue) and BET (red) isotherms
The derivation of the formula is more complicated than Langmuir's (see links for complete derivation). We obtain:
where x is the pressure divided by the vapor pressure for the adsorbate at that temperature (usually denoted ), v is the STP volume of adsorbed adsorbate, vmon is the STP volume of the amount of adsorbate required to form a monolayer, and c is the equilibrium constant K we used in Langmuir isotherm multiplied by the vapor pressure of the adsorbate. The key assumption used in deriving the BET equation that the successive heats of adsorption for all layers except the first are equal to the heat of condensation of the adsorbate.
The Langmuir isotherm is usually better for chemisorption, and the BET isotherm works better for physisorption for non-microporous surfaces.
Kisliuk
Two adsorbate nitrogen molecules adsorbing onto a tungsten adsorbent from the precursor state around an island of previously adsorbed adsorbate (left) and via random adsorption (right)
In other instances, molecular interactions between gas molecules previously adsorbed on a solid surface form significant interactions with gas molecules in the gaseous phases. Hence, adsorption of gas molecules to the surface is more likely to occur around gas molecules that are already present on the solid surface, rendering the Langmuir adsorption isotherm ineffective for the purposes of modelling. This effect was studied in a system where nitrogen was the adsorbate and tungsten was the adsorbent by Paul Kisliuk (1922–2008) in 1957.[16] To compensate for the increased probability of adsorption occurring around molecules present on the substrate surface, Kisliuk developed the precursor state theory, whereby molecules would enter a precursor state at the interface between the solid adsorbent and adsorbate in the gaseous phase. From here, adsorbate molecules would either adsorb to the adsorbent or desorb into the gaseous phase. The probability of adsorption occurring from the precursor state is dependent on the adsorbate's proximity to other adsorbate molecules that have already been adsorbed. If the adsorbate molecule in the precursor state is in close proximity to an adsorbate molecule that has already formed on the surface, it has a sticking probability reflected by the size of the SE constant and will either be adsorbed from the precursor state at a rate of kEC or will desorb into the gaseous phase at a rate of kES. If an adsorbate molecule enters the precursor state at a location that is remote from any other previously adsorbed adsorbate molecules, the sticking probability is reflected by the size of the SD constant.
These factors were included as part of a single constant termed a "sticking coefficient", kE, described below:
As SD is dictated by factors that are taken into account by the Langmuir model, SD can be assumed to be the adsorption rate constant. However, the rate constant for the Kisliuk model (R’) is different from that of the Langmuir model, as R’ is used to represent the impact of diffusion on monolayer formation and is proportional to the square root of the system's diffusion coefficient. The Kisliuk adsorption isotherm is written as follows, where θ(t) is fractional coverage of the adsorbent with adsorbate, and t is immersion time:
As can be seen in the formula, the variation of K must be isosteric, that is, at constant coverage. If we start from the BET isotherm and assume that the entropy change is the same for liquefaction and adsorption, we obtain
that is to say, adsorption is more exothermic than liquefaction.
Single-molecule explanation
The adsorption of ensemble molecules on a surface or interface can be divided into two processes: adsorption and desorption. If the adsorption rate wins the desorption rate, the molecules will accumulate over time giving the adsorption curve over time. If the desorption rate is larger, the number of molecules on the surface will decrease over time. The adsorption rate is dependent on the temperature, the diffusion rate of the solute (related to mean free path for pure gas), and the energy barrier between the molecule and the surface. The diffusion and key elements of the adsorption rate can be calculated using Fick's laws of diffusion and Einstein relation (kinetic theory). Under ideal conditions, when there is no energy barrier and all molecules that diffuse and collide with the surface get adsorbed, the number of molecules adsorbed at a surface of area on an infinite area surface can be directly integrated from Fick's second law differential equation to be:[17]
where is the surface area (unit m2), is the number concentration of the molecule in the bulk solution (unit #/m3), is the diffusion constant (unit m2/s), and is time (unit s). Further simulations and analysis of this equation[18] show that the square root dependence on the time is originated from the decrease of the concentrations near the surface under ideal adsorption conditions. Also, this equation only works for the beginning of the adsorption when a well-behaved concentration gradient forms near the surface. Correction on the reduction of the adsorption area and slowing down of the concentration gradient evolution have to be considered over a longer time.[19] Under real experimental conditions, the flow and the small adsorption area always make the adsorption rate faster than what this equation predicted, and the energy barrier will either accelerate this rate by surface attraction or slow it down by surface repulsion. Thus, the prediction from this equation is often a few to several orders of magnitude away from the experimental results. Under special cases, such as a very small adsorption area on a large surface, and under chemical equilibrium when there is no concentration gradience near the surface, this equation becomes useful to predict the adsorption rate with debatable special care to determine a specific value of in a particular measurement.[18]
The desorption of a molecule from the surface depends on the binding energy of the molecule to the surface and the temperature. The typical overall adsorption rate is thus often a combined result of the adsorption and desorption.
Quantum mechanical – thermodynamic modelling for surface area and porosity
Since 1980 two theories were worked on to explain adsorption and obtain equations that work. These two are referred to as the chi hypothesis, the quantum mechanical derivation, and excess surface work (ESW).[20] Both these theories yield the same equation for flat surfaces:
where U is the unit step function. The definitions of the other symbols is as follows:
where "ads" stands for "adsorbed", "m" stands for "monolayer equivalence" and "vap" is reference to the vapor pressure of the liquid adsorptive at the same temperature as the solid sample. The unit function creates the definition of the molar energy of adsorption for the first adsorbed molecule by:
The plot of adsorbed versus is referred to as the chi plot. For flat surfaces, the slope of the chi plot yields the surface area. Empirically, this plot was noticed as being a very good fit to the isotherm by Michael Polanyi[21][22][23] and also by Jan Hendrik de Boer and Cornelis Zwikker[24] but not pursued. This was due to criticism in the former case by Albert Einstein and in the latter case by Brunauer. This flat surface equation may be used as a "standard curve" in the normal tradition of comparison curves, with the exception that the porous sample's early portion of the plot of versus acts as a self-standard. Ultramicroporous, microporous and mesoporous conditions may be analyzed using this technique. Typical standard deviations for full isotherm fits including porous samples are less than 2%.
Notice that in this description of physical adsorption, the entropy of adsorption is consistent with the Dubinin thermodynamic criterion, that is the entropy of adsorption from the liquid state to the adsorbed state is approximately zero.
Adsorbents
Characteristics and general requirements
Activated carbon is used as an adsorbent
Adsorbents are used usually in the form of spherical pellets, rods, moldings, or monoliths with a hydrodynamic radius between 0.25 and 5mm. They must have high abrasion resistance, high thermal stability and small pore diameters, which results in higher exposed surface area and hence high capacity for adsorption. The adsorbents must also have a distinct pore structure that enables fast transport of the gaseous vapors.[25] Most industrial adsorbents fall into one of three classes:
Oxygen-containing compounds – Are typically hydrophilic and polar, including materials such as silica gel, limestone (calcium carbonate)[26] and zeolites.
Carbon-based compounds – Are typically hydrophobic and non-polar, including materials such as activated carbon and graphite.
Polymer-based compounds – Are polar or non-polar, depending on the functional groups in the polymer matrix.
Silica gel
Silica gel adsorber for NO2, Fixed Nitrogen Research Laboratory, ca.1930s
Silica gel is a chemically inert, non-toxic, polar and dimensionally stable (< 400°C or 750°F) amorphous form of SiO2. It is prepared by the reaction between sodium silicate and acetic acid, which is followed by a series of after-treatment processes such as aging, pickling, etc. These after-treatment methods results in various pore size distributions.
Silica is used for drying of process air (e.g. oxygen, natural gas) and adsorption of heavy (polar) hydrocarbons from natural gas.
Zeolites
Zeolites are natural or synthetic crystalline aluminosilicates, which have a repeating pore network and release water at high temperature. Zeolites are polar in nature.
They are manufactured by hydrothermal synthesis of sodium aluminosilicate or another silica source in an autoclave followed by ion exchange with certain cations (Na+, Li+, Ca2+, K+, NH4+). The channel diameter of zeolite cages usually ranges from 2 to 9 Å. The ion exchange process is followed by drying of the crystals, which can be pelletized with a binder to form macroporous pellets.
Zeolites are applied in drying of process air, CO2 removal from natural gas, CO removal from reforming gas, air separation, catalytic cracking, and catalytic synthesis and reforming.
Non-polar (siliceous) zeolites are synthesized from aluminum-free silica sources or by dealumination of aluminum-containing zeolites. The dealumination process is done by treating the zeolite with steam at elevated temperatures, typically greater than 500°C (930°F). This high temperature heat treatment breaks the aluminum-oxygen bonds and the aluminum atom is expelled from the zeolite framework.
Activated carbon
Activated carbon is a highly porous, amorphous solid consisting of microcrystallites with a graphite lattice, usually prepared in small pellets or a powder. It is non-polar and cheap. One of its main drawbacks is that it reacts with oxygen at moderate temperatures (over 300°C).
Activated carbon nitrogen isotherm showing a marked microporous type I behavior
Activated carbon can be manufactured from carbonaceous material, including coal (bituminous, subbituminous, and lignite), peat, wood, or nutshells (e.g., coconut). The manufacturing process consists of two phases, carbonization and activation.[27][28] The carbonization process includes drying and then heating to separate by-products, including tars and other hydrocarbons from the raw material, as well as to drive off any gases generated. The process is completed by heating the material over 400°C (750°F) in an oxygen-free atmosphere that cannot support combustion. The carbonized particles are then "activated" by exposing them to an oxidizing agent, usually steam or carbon dioxide at high temperature. This agent burns off the pore blocking structures created during the carbonization phase and so, they develop a porous, three-dimensional graphite lattice structure. The size of the pores developed during activation is a function of the time that they spend in this stage. Longer exposure times result in larger pore sizes. The most popular aqueous phase carbons are bituminous based because of their hardness, abrasion resistance, pore size distribution, and low cost, but their effectiveness needs to be tested in each application to determine the optimal product.
Activated carbon is used for adsorption of organic substances[29] and non-polar adsorbates and it is also usually used for waste gas (and waste water) treatment. It is the most widely used adsorbent since most of its chemical (e.g. surface groups) and physical properties (e.g. pore size distribution and surface area) can be tuned according to what is needed.[30] Its usefulness also derives from its large micropore (and sometimes mesopore) volume and the resulting high surface area. Recent research works reported activated carbon as an effective agent to adsorb cationic species of toxic metals from multi-pollutant systems and also proposed possible adsorption mechanisms with supporting evidences.[31]
Water adsorption
The adsorption of water at surfaces is of broad importance in chemical engineering, materials science and catalysis. Also termed surface hydration, the presence of physically or chemically adsorbed water at the surfaces of solids plays an important role in governing interface properties, chemical reaction pathways and catalytic performance in a wide range of systems. In the case of physically adsorbed water, surface hydration can be eliminated simply through drying at conditions of temperature and pressure allowing full vaporization of water. For chemically adsorbed water, hydration may be in the form of either dissociative adsorption, where H2O molecules are dissociated into surface adsorbed -H and -OH, or molecular adsorption (associative adsorption) where individual water molecules remain intact [32]
Adsorption solar heating and storage
The low cost ($200/ton) and high cycle rate (2,000 ×) of synthetic zeolites such as Linde 13X with water adsorbate has garnered much academic and commercial interest recently for use for thermal energy storage (TES), specifically of low-grade solar and waste heat. Several pilot projects have been funded in the EU from 2000 to the present (2020).[citation needed] The basic concept is to store solar thermal energy as chemical latent energy in the zeolite. Typically, hot dry air from flat plate solar collectors is made to flow through a bed of zeolite such that any water adsorbate present is driven off. Storage can be diurnal, weekly, monthly, or even seasonal depending on the volume of the zeolite and the area of the solar thermal panels. When heat is called for during the night, or sunless hours, or winter, humidified air flows through the zeolite. As the humidity is adsorbed by the zeolite, heat is released to the air and subsequently to the building space. This form of TES, with specific use of zeolites, was first taught by John Guerra in 1978.[33]
Carbon capture and storage
Typical adsorbents proposed for carbon capture and storage are zeolites and MOFs.[34] The customization of adsorbents makes them a potentially attractive alternative to absorption. Because adsorbents can be regenerated by temperature or pressure swing, this step can be less energy intensive than absorption regeneration methods.[35] Major problems that are present with adsorption cost in carbon capture are: regenerating the adsorbent, mass ratio, solvent/MOF, cost of adsorbent, production of the adsorbent, lifetime of adsorbent.[36]
In sorption enhanced water gas shift (SEWGS) technology a pre-combustion carbon capture process, based on solid adsorption, is combined with the water gas shift reaction (WGS) in order to produce a high pressure hydrogen stream.[37] The CO2 stream produced can be stored or used for other industrial processes.[38]
Protein adsorption is a process that has a fundamental role in the field of biomaterials. Indeed, biomaterial surfaces in contact with biological media, such as blood or serum, are immediately coated by proteins. Therefore, living cells do not interact directly with the biomaterial surface, but with the adsorbed proteins layer. This protein layer mediates the interaction between biomaterials and cells, translating biomaterial physical and chemical properties into a "biological language".[39] In fact, cell membranereceptors bind to protein layer bioactive sites and these receptor-protein binding events are transduced, through the cell membrane, in a manner that stimulates specific intracellular processes that then determine cell adhesion, shape, growth and differentiation. Protein adsorption is influenced by many surface properties such as surface wettability, surface chemical composition [40] and surface nanometre-scale morphology.[41] Surfactant adsorption is a similar phenomenon, but utilising surfactant molecules in the place of proteins.[42]
Adsorption chillers
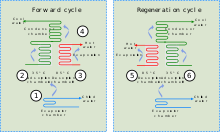
A schematic diagram of an adsorption chiller: (1) heat is lost through evaporation of refrigerant, (2) refrigerant vapour is adsorbed onto the solid medium, (3) refrigerant is desorbed from the solid medium section not in use, (4) refrigerant is condensed and returned to the start, (5) & (6) solid medium is cycled between adsorption and desorption to regenerate it.
Combining an adsorbent with a refrigerant, adsorption chillers use heat to provide a cooling effect. This heat, in the form of hot water, may come from any number of industrial sources including waste heat from industrial processes, prime heat from solar thermal installations or from the exhaust or water jacket heat of a piston engine or turbine.
Although there are similarities between adsorption chillers and absorption refrigeration, the former is based on the interaction between gases and solids. The adsorption chamber of the chiller is filled with a solid material (for example zeolite, silica gel, alumina, active carbon or certain types of metal salts), which in its neutral state has adsorbed the refrigerant. When heated, the solid desorbs (releases) refrigerant vapour, which subsequently is cooled and liquefied. This liquid refrigerant then provides a cooling effect at the evaporator from its enthalpy of vaporization. In the final stage the refrigerant vapour is (re)adsorbed into the solid.[43] As an adsorption chiller requires no compressor, it is relatively quiet.
Portal site mediated adsorption
Portal site mediated adsorption is a model for site-selective activated gas adsorption in metallic catalytic systems that contain a variety of different adsorption sites. In such systems, low-coordination "edge and corner" defect-like sites can exhibit significantly lower adsorption enthalpies than high-coordination (basal plane) sites. As a result, these sites can serve as "portals" for very rapid adsorption to the rest of the surface. The phenomenon relies on the common "spillover" effect (described below), where certain adsorbed species exhibit high mobility on some surfaces. The model explains seemingly inconsistent observations of gas adsorption thermodynamics and kinetics in catalytic systems where surfaces can exist in a range of coordination structures, and it has been successfully applied to bimetallic catalytic systems where synergistic activity is observed.
In contrast to pure spillover, portal site adsorption refers to surface diffusion to adjacent adsorption sites, not to non-adsorptive support surfaces.
The model appears to have been first proposed for carbon monoxide on silica-supported platinum by Brandt et al. (1993).[44] A similar, but independent model was developed by King and co-workers[45][46][47] to describe hydrogen adsorption on silica-supported alkali promoted ruthenium, silver-ruthenium and copper-ruthenium bimetallic catalysts. The same group applied the model to CO hydrogenation (Fischer–Tropsch synthesis).[48] Zupanc et al. (2002) subsequently confirmed the same model for hydrogen adsorption on magnesia-supported caesium-ruthenium bimetallic catalysts.[49] Trens et al. (2009) have similarly described CO surface diffusion on carbon-supported Pt particles of varying morphology.[50]
Adsorption spillover
In the case catalytic or adsorbent systems where a metal species is dispersed upon a support (or carrier) material (often quasi-inert oxides, such as alumina or silica), it is possible for an adsorptive species to indirectly adsorb to the support surface under conditions where such adsorption is thermodynamically unfavorable. The presence of the metal serves as a lower-energy pathway for gaseous species to first adsorb to the metal and then diffuse on the support surface. This is possible because the adsorbed species attains a lower energy state once it has adsorbed to the metal, thus lowering the activation barrier between the gas phase species and the support-adsorbed species.
Hydrogen spillover is the most common example of an adsorptive spillover. In the case of hydrogen, adsorption is most often accompanied with dissociation of molecular hydrogen (H2) to atomic hydrogen (H), followed by spillover of the hydrogen atoms present.
The spillover effect has been used to explain many observations in heterogeneous catalysis and adsorption.[51]
Adsorption of molecules onto polymer surfaces is central to a number of applications, including development of non-stick coatings and in various biomedical devices. Polymers may also be adsorbed to surfaces through polyelectrolyte adsorption.
In viruses
Adsorption is the first step in the viral life cycle. The next steps are penetration, uncoating, synthesis (transcription if needed, and translation), and release. The virus replication cycle, in this respect, is similar for all types of viruses. Factors such as transcription may or may not be needed if the virus is able to integrate its genomic information in the cell's nucleus, or if the virus can replicate itself directly within the cell's cytoplasm.
In popular culture
The game of Tetris is a puzzle game in which blocks of 4 are adsorbed onto a surface during game play. Scientists have used Tetris blocks "as a proxy for molecules with a complex shape" and their "adsorption on a flat surface" for studying the thermodynamics of nanoparticles.[52][53]
The Haber process, also called the Haber–Bosch process, is the main industrial procedure for the production of ammonia. The German chemists Fritz Haber and Carl Bosch developed it in the first decade of the 20th century. The process converts atmospheric nitrogen (N2) to ammonia (NH3) by a reaction with hydrogen (H2) using an iron metal catalyst under high temperatures and pressures. This reaction is slightly exothermic (i.e. it releases energy), meaning that the reaction is favoured at lower temperatures and higher pressures. It decreases entropy, complicating the process. Hydrogen is produced via steam reforming, followed by an iterative closed cycle to react hydrogen with nitrogen to produce ammonia.
Chemisorption is a kind of adsorption which involves a chemical reaction between the surface and the adsorbate. New chemical bonds are generated at the adsorbent surface. Examples include macroscopic phenomena that can be very obvious, like corrosion, and subtler effects associated with heterogeneous catalysis, where the catalyst and reactants are in different phases. The strong interaction between the adsorbate and the substrate surface creates new types of electronic bonds.
Physisorption, also called physical adsorption, is a process in which the electronic structure of the atom or molecule is barely perturbed upon adsorption.
Temperature programmed desorption (TPD) is the method of observing desorbed molecules from a surface when the surface temperature is increased. When experiments are performed using well-defined surfaces of single-crystalline samples in a continuously pumped ultra-high vacuum (UHV) chamber, then this experimental technique is often also referred to as thermal desorption spectroscopy or thermal desorption spectrometry (TDS).
Heterogeneous catalysis is catalysis where the phase of catalysts differs from that of the reactants or products. The process contrasts with homogeneous catalysis where the reactants, products and catalyst exist in the same phase. Phase distinguishes between not only solid, liquid, and gas components, but also immiscible mixtures, or anywhere an interface is present.
Desorption is the physical process where adsorbed atoms or molecules are released from a surface into the surrounding vacuum or fluid. This occurs when a molecule gains enough energy to overcome the activation barrier and the binding energy that keep it attached to the surface.
Gas mixtures can be effectively separated by synthetic membranes made from polymers such as polyamide or cellulose acetate, or from ceramic materials.
Brunauer–Emmett–Teller (BET) theory aims to explain the physical adsorption of gas molecules on a solid surface and serves as the basis for an important analysis technique for the measurement of the specific surface area of materials. The observations are very often referred to as physical adsorption or physisorption. In 1938, Stephen Brunauer, Paul Hugh Emmett, and Edward Teller presented their theory in the Journal of the American Chemical Society. BET theory applies to systems of multilayer adsorption that usually utilizes a probing gas (called the adsorbate) that does not react chemically with the adsorptive (the material upon which the gas attaches to) to quantify specific surface area. Nitrogen is the most commonly employed gaseous adsorbate for probing surface(s). For this reason, standard BET analysis is most often conducted at the boiling temperature of N2 (77 K). Other probing adsorbates are also utilized, albeit less often, allowing the measurement of surface area at different temperatures and measurement scales. These include argon, carbon dioxide, and water. Specific surface area is a scale-dependent property, with no single true value of specific surface area definable, and thus quantities of specific surface area determined through BET theory may depend on the adsorbate molecule utilized and its adsorption cross section.
The sticking probability is the probability that molecules are trapped on surfaces and adsorb chemically. From Langmuir's adsorption isotherm, molecules cannot adsorb on surfaces when the adsorption sites are already occupied by other molecules, so the sticking probability can be expressed as follows:
Pressure swing adsorption (PSA) is a technique used to separate some gas species from a mixture of gases under pressure according to the species' molecular characteristics and affinity for an adsorbent material. It operates at near-ambient temperature and significantly differs from the cryogenic distillation commonly used to separate gases. Selective adsorbent materials are used as trapping material, preferentially adsorbing the target gas species at high pressure. The process then swings to low pressure to desorb the adsorbed gas.
Reactions on surfaces are reactions in which at least one of the steps of the reaction mechanism is the adsorption of one or more reactants. The mechanisms for these reactions, and the rate equations are of extreme importance for heterogeneous catalysis. Via scanning tunneling microscopy, it is possible to observe reactions at the solid gas interface in real space, if the time scale of the reaction is in the correct range. Reactions at the solid–gas interface are in some cases related to catalysis.
The Freundlich equation or Freundlich adsorption isotherm, an adsorption isotherm, is an empirical relationship between the quantity of a gas adsorbed into a solid surface and the gas pressure. The same relationship is also applicable for the concentration of a solute adsorbed onto the surface of a solid and the concentration of the solute in the liquid phase. In 1909, Herbert Freundlich gave an expression representing the isothermal variation of adsorption of a quantity of gas adsorbed by unit mass of solid adsorbent with gas pressure. This equation is known as Freundlich adsorption isotherm or Freundlich adsorption equation. As this relationship is entirely empirical, in the case where adsorption behavior can be properly fit by isotherms with a theoretical basis, it is usually appropriate to use such isotherms instead. The Freundlich equation is also derived (non-empirically) by attributing the change in the equilibrium constant of the binding process to the heterogeneity of the surface and the variation in the heat of adsorption.
Sticking coefficient is the term used in surface physics to describe the ratio of the number of adsorbate atoms that adsorb, or "stick", to a surface to the total number of atoms that impinge upon that surface during the same period of time. Sometimes the symbol Sc is used to denote this coefficient, and its value is between 1 and 0. The coefficient is a function of surface temperature, surface coverage (θ) and structural details as well as the kinetic energy of the impinging particles. The original formulation was for molecules adsorbing from the gas phase and the equation was later extended to adsorption from the liquid phase by comparison with molecular dynamics simulations. For use in adsorption from liquids the equation is expressed based on solute density rather than the pressure.
The Langmuir adsorption model explains adsorption by assuming an adsorbate behaves as an ideal gas at isothermal conditions. According to the model, adsorption and desorption are reversible processes. This model even explains the effect of pressure i.e. at these conditions the adsorbate's partial pressure is related to its volume V adsorbed onto a solid adsorbent. The adsorbent, as indicated in the figure, is assumed to be an ideal solid surface composed of a series of distinct sites capable of binding the adsorbate. The adsorbate binding is treated as a chemical reaction between the adsorbate gaseous molecule and an empty sorption site S. This reaction yields an adsorbed species with an associated equilibrium constant :
In materials science and biology, capillary condensation is the "process by which multilayer adsorption from the vapor [phase] into a porous medium proceeds to the point at which pore spaces become filled with condensed liquid from the vapor [phase]." The unique aspect of capillary condensation is that vapor condensation occurs below the saturation vapor pressure, Psat, of the pure liquid. This result is due to an increased number of van der Waals interactions between vapor phase molecules inside the confined space of a capillary. Once condensation has occurred, a meniscus immediately forms at the liquid-vapor interface which allows for equilibrium below the saturation vapor pressure. Meniscus formation is dependent on the surface tension of the liquid and the shape of the capillary, as shown by the Young-Laplace equation. As with any liquid-vapor interface involving a meniscus, the Kelvin equation provides a relation for the difference between the equilibrium vapor pressure and the saturation vapor pressure. A capillary does not necessarily have to be a tubular, closed shape, but can be any confined space with respect to its surroundings.
Supercritical adsorption also referred to as the adsorption of supercritical fluids, is the adsorption at above-critical temperatures. There are different tacit understandings of supercritical fluids. For example, “a fluid is considered to be ‘supercritical’ when its temperature and pressure exceed the temperature and pressure at the critical point”. In the studies of supercritical extraction, however, “supercritical fluid” is applied for a narrow temperature region of 1-1.2 or to +10 K, which is called the supercritical region.
Adsorption is the adhesion of ions or molecules onto the surface of another phase. Adsorption may occur via physisorption and chemisorption. Ions and molecules can adsorb to many types of surfaces including polymer surfaces. A polymer is a large molecule composed of repeating subunits bound together by covalent bonds. In dilute solution, polymers form globule structures. When a polymer adsorbs to a surface that it interacts favorably with, the globule is essentially squashed, and the polymer has a pancake structure.
Silanization of silicon and mica is the coating of these materials with a thin layer of self assembling units.
The Henry adsorption constant is the constant appearing in the linear adsorption isotherm, which formally resembles Henry's law; therefore, it is also called Henry's adsorption isotherm. It is named after British chemist William Henry. This is the simplest adsorption isotherm in that the amount of the surface adsorbate is represented to be proportional to the partial pressure of the adsorptive gas:
The potential theory of Polanyi, also called Polanyi adsorption potential theory, is a model of adsorption proposed by Michael Polanyi where adsorption can be measured through the equilibrium between the chemical potential of a gas near the surface and the chemical potential of the gas from a large distance away. In this model, he assumed that the attraction largely due to Van Der Waals forces of the gas to the surface is determined by the position of the gas particle from the surface, and that the gas behaves as an ideal gas until condensation where the gas exceeds its equilibrium vapor pressure. While the adsorption theory of Henry is more applicable in low pressure and BET adsorption isotherm equation is more useful at from 0.05 to 0.35 P/Po, the Polanyi potential theory has much more application at higher P/Po (~0.1–0.8).
References
↑ Guruge, Amila Ruwan (2021-02-17). "Absorption Vs Adsorption". Chemical and Process Engineering. Retrieved 2023-11-26.
↑ "Glossary". The Brownfields and Land Revitalization Technology Support Center. Archived from the original on 2008-02-18. Retrieved 2009-12-21.
↑ Czelej, K.; Cwieka, K.; Kurzydlowski, K.J. (May 2016). "CO2 stability on the Ni low-index surfaces: Van der Waals corrected DFT analysis". Catalysis Communications. 80 (5): 33–38. doi:10.1016/j.catcom.2016.03.017.
↑ Czelej, K.; Cwieka, K.; Colmenares, J.C.; Kurzydlowski, K.J. (2016). "Insight on the Interaction of Methanol-Selective Oxidation Intermediates with Au- or/and Pd-Containing Monometallic and Bimetallic Core@Shell Catalysts". Langmuir. 32 (30): 7493–7502. doi:10.1021/acs.langmuir.6b01906. PMID27373791.
↑ Kayser, Heinrich (1881). "Über die Verdichtung von Gasen an Oberflächen in ihrer Abhängigkeit von Druck und Temperatur". Annalen der Physik und Chemie. 248 (4): 526–537. Bibcode:1881AnP...248..526K. doi:10.1002/andp.18812480404.. In this study of the adsorption of gases by charcoal, the first use of the word "adsorption" appears on page 527: "Schon Saussure kannte die beiden für die Grösse der Adsorption massgebenden Factoren, den Druck und die Temperatur, da er Erniedrigung des Druckes oder Erhöhung der Temperatur zur Befreiung der porösen Körper von Gasen benutzte." ("Saussaure already knew the two factors that determine the quantity of adsorption – [namely,] the pressure and temperature – since he used the lowering of the pressure or the raising of the temperature to free the porous substances of gases.")
1 2 Burke GM, Wurster DE, Buraphacheep V, Berg MJ, Veng-Pedersen P, Schottelius DD. Model selection for the adsorption of phenobarbital by activated charcoal. Pharm Res. 1991;8(2):228‐231. doi:10.1023/a:1015800322286
↑ Physical Chemistry of Surfaces. Arthur W. Adamson. Interscience (Wiley), New York 6th ed
↑ Langmuir, I.; Schaefer, V.J. (1937). "The Effect of Dissolved Salts on Insoluble Monolayers". Journal of the American Chemical Society. 29 (11): 2400–2414. doi:10.1021/ja01290a091.
↑ Ward, A.F.H.; Tordai, L. (1946). "Time-dependence of Boundary Tensions of Solutions I. The Role of Diffusion in Time-effects". Journal of Chemical Physics. 14 (7): 453–461. Bibcode:1946JChPh..14..453W. doi:10.1063/1.1724167.
↑ Condon, James (2020). Surface Area and Porosity Determinations by Physisorption, Measurement, Classical Theory and Quantum Theory, 2nd edition. Amsterdam.NL: Elsevier. pp.Chapters 3, 4 and 5. ISBN978-0-12-818785-2.
↑ Polanyi, M. (1914). "Über die Adsorption vom Standpunkt des dritten Wärmesatzes". Verhandlungen der Deutschen Physikalischen Gesellschaft (in German). 16: 1012.
↑ Polanyi, M. (1920). "Neueres über Adsorption und Ursache der Adsorptionskräfte". Zeitschrift für Elektrochemie. 26: 370–374.
↑ Polanyi, M. (1929). "Grundlagen der Potentialtheorie der Adsorption". Zeitschrift für Elektrochemie (in German). 35: 431–432.
↑ deBoer, J.H.; Zwikker, C. (1929). "Adsorption als Folge von Polarisation". Zeitschrift für Physikalische Chemie (in German). B3: 407–420.
↑ Viswambari Devi, R; Nair, Vijay V; Sathyamoorthy, P; Doble, Mukesh (2022). "Mixture of CaCO3 Polymorphs Serves as Best Adsorbent of Heavy Metals in Quadruple System". Journal of Hazardous, Toxic & Radioactive Waste. 26 (1). doi:10.1061/(ASCE)HZ.2153-5515.0000651. S2CID240454883.
↑ Spessato, L. et al. KOH-super activated carbon from biomass waste: Insights into the paracetamol adsorption mechanism and thermal regeneration cycles. Journal of Hazardous Materials, Vol. 371, Pages 499-505, 2019.
↑ Spessato, L. et al. Optimization of Sibipiruna activated carbon preparation by simplex-centroid mixture design for simultaneous adsorption of rhodamine B and metformin. Journal of Hazardous Materials, Vol. 411, Page 125166, 2021.
↑ Malhotra, Milan; Suresh, Sumathi; Garg, Anurag (2018). "Tea waste derived activated carbon for the adsorption of sodium diclofenac from wastewater: adsorbent characteristics, adsorption isotherms, kinetics, and thermodynamics". Environmental Science and Pollution Research. 25 (32): 32210–32220. Bibcode:2018ESPR...2532210M. doi:10.1007/s11356-018-3148-y. PMID30221322. S2CID52280860.
↑ Mohan, S; Nair, Vijay V (2020). "Comparative study of separation of heavy metals from leachate using activated carbon and fuel ash". Journal of Hazardous, Toxic & Radioactive Waste. 24 (4): 473–491. doi:10.1061/(ASCE)HZ.2153-5515.0000520. PMID04020031. S2CID219747988.
↑ U.S. Pat. No. 4,269,170, "Adsorption solar heating and storage"; Inventor: John M. Guerra; Granted May 26, 1981
↑ Berend, Smit; Reimer, Jeffery A; Oldenburg, Curtis M; Bourg, Ian C (2014). Introduction to carbon capture and sequestration. Imperial College Press. ISBN9781306496834.
↑ Sathre, Roger; Masanet, Eric (2013-03-18). "Prospective life-cycle modeling of a carbon capture and storage system using metal–organic frameworks for CO2 capture". RSC Advances. 3 (15): 4964. Bibcode:2013RSCAd...3.4964S. doi:10.1039/C3RA40265G. ISSN2046-2069.
↑ (Eric) van Dijk, H. A. J.; Cobden, Paul D.; Lukashuk, Liliana; de Water, Leon van; Lundqvist, Magnus; Manzolini, Giampaolo; Cormos, Calin-Cristian; van Dijk, Camiel; Mancuso, Luca; Johns, Jeremy; Bellqvist, David (1 October 2018). "STEPWISE Project: Sorption-Enhanced Water-Gas Shift Technology to Reduce Carbon Footprint in the Iron and Steel Industry". Johnson Matthey Technology Review. 62 (4): 395–402. doi:10.1595/205651318X15268923666410. hdl:11311/1079169. S2CID139928989.
↑ Wilson, CJ; Clegg, RE; Leavesley, DI; Pearcy, MJ (2005). "Mediation of Biomaterial-Cell Interactions by Adsorbed Proteins: A Review". Tissue Engineering. 11 (1): 1–18. doi:10.1089/ten.2005.11.1. PMID15738657. S2CID19306269.
↑ Pilatowsky, I.; Romero, R.J.; Isaza, C.A.; Gamboa, S.A.; Sebastian, P.J.; Rivera, W. (2011). "Sorption Refrigeration Systems". Cogeneration Fuel Cell-Sorption Air Conditioning Systems. Green Energy and Technology. Springer. pp.99, 100. doi:10.1007/978-1-84996-028-1_5. ISBN978-1-84996-027-4.
↑ Brandt, Robert K.; Hughes, M.R.; Bourget, L.P.; Truszkowska, K.; Greenler, Robert G. (April 1993). "The interpretation of CO adsorbed on Pt/SiO2 of two different particle-size distributions". Surface Science. 286 (1–2): 15–25. Bibcode:1993SurSc.286...15B. doi:10.1016/0039-6028(93)90552-U.
↑ Uner, D.O.; Savargoankar, N.; Pruski, M.; King, T.S. (1997). "The effects of alkali promoters on the dynamics of hydrogen chemisorption and syngas reaction kinetics on Ru/SiO2 surfaces". Dynamics of Surfaces and Reaction Kinetics in Heterogeneous Catalysis, Proceedings of the International Symposium. Studies in Surface Science and Catalysis. Vol.109. pp.315–324. doi:10.1016/s0167-2991(97)80418-1. ISBN9780444826091.
↑ Narayan, R.L; King, T.S (March 1998). "Hydrogen adsorption states on silica-supported Ru–Ag and Ru–Cu bimetallic catalysts investigated via microcalorimetry". Thermochimica Acta. 312 (1–2): 105–114. doi:10.1016/S0040-6031(97)00444-9.
↑ Uner, D. O. (1 June 1998). "A Sensible Mechanism of Alkali Promotion in Fischer−Tropsch Synthesis: Adsorbate Mobilities". Industrial & Engineering Chemistry Research. 37 (6): 2239–2245. doi:10.1021/ie970696d.
↑ Zupanc, C.; Hornung, A.; Hinrichsen, O.; Muhler, M. (July 2002). "The Interaction of Hydrogen with Ru/MgO Catalysts". Journal of Catalysis. 209 (2): 501–514. doi:10.1006/jcat.2002.3647.
↑ Trens, Philippe; Durand, Robert; Coq, Bernard; Coutanceau, Christophe; Rousseau, Séverine; Lamy, Claude (November 2009). "Poisoning of Pt/C catalysts by CO and its consequences over the kinetics of hydrogen chemisorption". Applied Catalysis B: Environmental. 92 (3–4): 280–284. Bibcode:2009AppCB..92..280T. doi:10.1016/j.apcatb.2009.08.004.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.