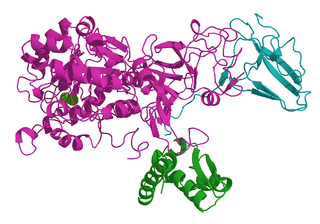
Ureases, functionally, belong to the superfamily of amidohydrolases and phosphotriesterases. Ureases are found in numerous bacteria, fungi, algae, plants, and some invertebrates, as well as in soils, as a soil enzyme. They are nickel-containing metalloenzymes of high molecular weight.
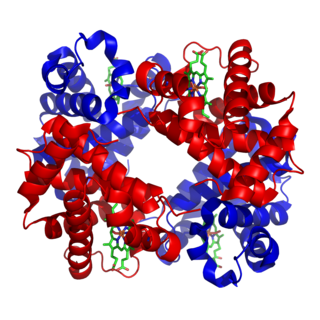
Metalloprotein is a generic term for a protein that contains a metal ion cofactor. A large proportion of all proteins are part of this category. For instance, at least 1000 human proteins contain zinc-binding protein domains although there may be up to 3000 human zinc metalloproteins.
Artificial photosynthesis is a chemical process that biomimics the natural process of photosynthesis to convert sunlight, water, and carbon dioxide into carbohydrates and oxygen. The term artificial photosynthesis is commonly used to refer to any scheme for capturing and storing the energy from sunlight in the chemical bonds of a fuel. Photocatalytic water splitting converts water into hydrogen and oxygen and is a major research topic of artificial photosynthesis. Light-driven carbon dioxide reduction is another process studied that replicates natural carbon fixation.
Iron–sulfur proteins are proteins characterized by the presence of iron–sulfur clusters containing sulfide-linked di-, tri-, and tetrairon centers in variable oxidation states. Iron–sulfur clusters are found in a variety of metalloproteins, such as the ferredoxins, as well as NADH dehydrogenase, hydrogenases, coenzyme Q – cytochrome c reductase, succinate – coenzyme Q reductase and nitrogenase. Iron–sulfur clusters are best known for their role in the oxidation-reduction reactions of electron transport in mitochondria and chloroplasts. Both Complex I and Complex II of oxidative phosphorylation have multiple Fe–S clusters. They have many other functions including catalysis as illustrated by aconitase, generation of radicals as illustrated by SAM-dependent enzymes, and as sulfur donors in the biosynthesis of lipoic acid and biotin. Additionally, some Fe–S proteins regulate gene expression. Fe–S proteins are vulnerable to attack by biogenic nitric oxide, forming dinitrosyl iron complexes. In most Fe–S proteins, the terminal ligands on Fe are thiolate, but exceptions exist.
A hydrogenase is an enzyme that catalyses the reversible oxidation of molecular hydrogen (H2), as shown below:
Bioorganometallic chemistry is the study of biologically active molecules that contain carbon directly bonded to metals or metalloids. The importance of main-group and transition-metal centers has long been recognized as important to the function of enzymes and other biomolecules. However, only a small subset of naturally-occurring metal complexes and synthetically prepared pharmaceuticals are organometallic; that is, they feature a direct covalent bond between the metal(loid) and a carbon atom. The first, and for a long time, the only examples of naturally occurring bioorganometallic compounds were the cobalamin cofactors (vitamin B12) in its various forms. In the 21st century, discovery of new systems containing carbon-metal bonds in biology, bioorganometallic chemistry is rapidly emerging as a distinct subdiscipline of bioinorganic chemistry that straddles organometallic chemistry and biochemistry. Naturally occurring bioorganometallics include enzymes and sensor proteins. Also within this realm are synthetically prepared organometallic compounds that serve as new drugs and imaging agents (technetium-99m sestamibi) as well as the principles relevant to the toxicology of organometallic compounds (e.g., methylmercury). Consequently, bioorganometallic chemistry is increasingly relevant to medicine and pharmacology.

Cupriavidus necator is a Gram-negative soil bacterium of the class Betaproteobacteria.
A hydrogenase mimic or bio-mimetic is an enzyme mimic of hydrogenases.
In chemistry, a (redox) non-innocent ligand is a ligand in a metal complex where the oxidation state is not clear. Typically, complexes containing non-innocent ligands are redox active at mild potentials. The concept assumes that redox reactions in metal complexes are either metal or ligand localized, which is a simplification, albeit a useful one.
In enzymology, carbon monoxide dehydrogenase (CODH) (EC 1.2.7.4) is an enzyme that catalyzes the chemical reaction
In enzymology, a cytochrome-c3 hydrogenase (EC 1.12.2.1) is an enzyme that catalyzes the chemical reaction

In enzymology, ferredoxin hydrogenase, also referred to as [Fe-Fe]hydrogenase, H2 oxidizing hydrogenase, H2 producing hydrogenase, bidirectional hydrogenase, hydrogenase (ferredoxin), hydrogenlyase, and uptake hydrogenase, is found in Clostridium pasteurianum, Clostridium acetobutylicum,Chlamydomonas reinhardtii, and other organisms. The systematic name of this enzyme is hydrogen:ferredoxin oxidoreductase
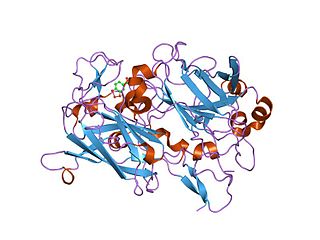
Dioxygenases are oxidoreductase enzymes. Aerobic life, from simple single-celled bacteria species to complex eukaryotic organisms, has evolved to depend on the oxidizing power of dioxygen in various metabolic pathways. From energetic adenosine triphosphate (ATP) generation to xenobiotic degradation, the use of dioxygen as a biological oxidant is widespread and varied in the exact mechanism of its use. Enzymes employ many different schemes to use dioxygen, and this largely depends on the substrate and reaction at hand.
An enzymatic biofuel cell is a specific type of fuel cell that uses enzymes as a catalyst to oxidize its fuel, rather than precious metals. Enzymatic biofuel cells, while currently confined to research facilities, are widely prized for the promise they hold in terms of their relatively inexpensive components and fuels, as well as a potential power source for bionic implants.

Iron–nickel (Fe–Ni) clusters are metal clusters consisting of iron and nickel, i.e. Fe–Ni structures displaying polyhedral frameworks held together by two or more metal–metal bonds per metal atom, where the metal atoms are located at the vertices of closed, triangulated polyhedra.
Hydrogenases are enzymes that catalyze the reversible activation of hydrogen and which occur widely in prokaryotes as well as in some eukaryotes. There are various types of hydrogenases, but all of them seem to contain at least one iron-sulphur cluster. They can be broadly divided into two groups: hydrogenases containing nickel and, in some cases, also selenium and those lacking nickel.

An iron hydride is a chemical system which contains iron and hydrogen in some associated form.

Nickel superoxide dismutase (Ni-SOD) is a metalloenzyme that, like the other superoxide dismutases, protects cells from oxidative damage by catalyzing the disproportionation of the cytotoxic superoxide radical to hydrogen peroxide and molecular oxygen. Superoxide is a reactive oxygen species that is produced in large amounts during photosynthesis and aerobic cellular respiration. The equation for the disproportionation of superoxide is shown below:

Acetyl-CoA synthase (ACS), not to be confused with Acetyl-CoA synthetase or Acetate-CoA ligase, is a nickel-containing enzyme involved in the metabolic processes of cells. Together with Carbon monoxide dehydrogenase (CODH), it forms the bifunctional enzyme Acetyl-CoA Synthase/Carbon Monoxide Dehydrogenase (ACS/CODH) found in anaerobic organisms such as archaea and bacteria. The ACS/CODH enzyme works primarily through the Wood–Ljungdahl pathway which converts carbon dioxide to Acetyl-CoA. The recommended name for this enzyme is CO-methylating acetyl-CoA synthase.

Wolfgang Lubitz is a German chemist and biophysicist. He is currently a director emeritus at the Max Planck Institute for Chemical Energy Conversion. He is well known for his work on bacterial photosynthetic reaction centres, hydrogenase enzymes, and the oxygen-evolving complex using a variety of biophysical techniques. He has been recognized by a Festschrift for his contributions to electron paramagnetic resonance (EPR) and its applications to chemical and biological systems.

![The structure of [NiFe] hydrogenase isolated from D. vulgaris Miyazaki F consists of two subunits: the large subunit (blue) and the small subunit (magenta). The Figure was prepared using Jmol and the coordinates from PDB: 1H2A . Desulfovibrio vulgaris Miyazaki F.jpg](http://upload.wikimedia.org/wikipedia/commons/thumb/6/69/Desulfovibrio_vulgaris_Miyazaki_F.jpg/300px-Desulfovibrio_vulgaris_Miyazaki_F.jpg)
![The active site of [NiFe] hydrogenase in the oxidized form. L refers to non-protein ligand (1 C[?]O and 2 C[?]N). X can be an oxide, sulfur, hydroperoxide, or a hydroxide. Nickel Iron Hydrogenase Active Site.png](http://upload.wikimedia.org/wikipedia/commons/thumb/e/e2/Nickel_Iron_Hydrogenase_Active_Site.png/150px-Nickel_Iron_Hydrogenase_Active_Site.png)
![Illustration of [NiFe] hydrogenase enzyme with three Fe-S clusters in the small subunit and with Mg and Ni-Fe dimetal active site in the large subunit. Figure was prepared using Jmol and the coordinates from PDB: 1H2A . Mg ion = neon green; Ni ion = dark green; Fe ion = orange; sulfur = yellow; oxygen = red; carbon = dark gray NiFe hydrogenase active site.jpg](http://upload.wikimedia.org/wikipedia/commons/thumb/d/d6/NiFe_hydrogenase_active_site.jpg/500px-NiFe_hydrogenase_active_site.jpg)
![Figure 5. Different redox states of [NiFe] hydrogenase's metal active site. The redox states in the red are the inactive redox states. The redox states in the green are the active redox states. (Adapted from ). Redox states of NiFe hydrogenase.png](http://upload.wikimedia.org/wikipedia/commons/thumb/a/a1/Redox_states_of_NiFe_hydrogenase.png/750px-Redox_states_of_NiFe_hydrogenase.png)
![Figure 6. Illustration of the [NiFe] hydrogenase active site inhibited by CO. Top-down view (left). Side view (center). Chemdraw depiction of the inhibited active site (right). The figure was prepared with Jmol and coordinates from 1UBK.pdb. Ni ion = green; Fe ion = orange; sulfur = yellow; oxygen = red; carbon = dark gray; nitrogen = blue. NiFe-CO inhibited.jpg](http://upload.wikimedia.org/wikipedia/commons/thumb/d/d7/NiFe-CO_inhibited.jpg/600px-NiFe-CO_inhibited.jpg)