
Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen due to a lower than normal number of red blood cells, a reduction in the amount of hemoglobin or hemoglobin abnormalities. The name is derived from Ancient Greek: ἀναιμία anaimia, meaning 'lack of blood', from ἀν- an-, 'not' and αἷμα haima, 'blood'. When anemia comes on slowly, the symptoms are often vague, such as tiredness, weakness, shortness of breath, headaches, and a reduced ability to exercise. When anemia is acute, symptoms may include confusion, feeling like one is going to pass out, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Symptoms of anemia depend on how quickly hemoglobin decreases. Additional symptoms may occur depending on the underlying cause. Preoperative anemia can increase the risk of needing a blood transfusion following surgery. Anemia can be temporary or long term and can range from mild to severe.

Mitochondrial disease is a group of disorders caused by mitochondrial dysfunction. Mitochondria are the organelles that generate energy for the cell and are found in every cell of the human body except red blood cells. They convert the energy of food molecules into the ATP that powers most cell functions.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.

Human mitochondrial genetics is the study of the genetics of human mitochondrial DNA. The human mitochondrial genome is the entirety of hereditary information contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
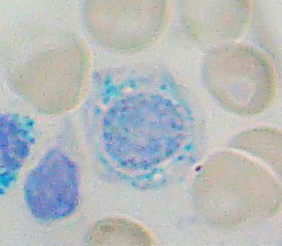
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.

Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and MDS/AML, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing and cognitive impairment can be a feature. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

Exocrine pancreatic insufficiency (EPI) is the inability to properly digest food due to a lack or reduction of digestive enzymes made by the pancreas. EPI can occur in humans and is prevalent in many conditions such as cystic fibrosis, Shwachman–Diamond syndrome, different types of pancreatitis, multiple types of diabetes mellitus, advanced renal disease, older adults, celiac disease, IBS-D, IBD, HIV, alcohol-related liver disease, Sjogren syndrome, tobacco use, and use of somatostatin analogues.
Reticulocytopenia is the medical term for an abnormal decrease in circulating red blood cell precursors (reticulocytes) that can lead to anemia due to resulting low red blood cell (erythrocyte) production. Reticulocytopenia may be an isolated finding or it may not be associated with abnormalities in other hematopoietic cell lineages such as those that produce white blood cells (leukocytes) or platelets (thrombocytes), a decrease in all three of these lineages is referred to as pancytopenia.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
Chronic progressive external ophthalmoplegia (CPEO) is a type of eye disorder characterized by slowly progressive inability to move the eyes and eyebrows. It is often the only feature of mitochondrial disease, in which case the term CPEO may be given as the diagnosis. In other people suffering from mitochondrial disease, CPEO occurs as part of a syndrome involving more than one part of the body, such as Kearns–Sayre syndrome. Occasionally CPEO may be caused by conditions other than mitochondrial diseases.
Congenital hypoplastic anemia is a congenital disorder that occasionally also includes leukopenia and thrombocytopenia and is characterized by deficiencies of red cell precursors.

Renal cysts and diabetes syndrome (RCAD), also known as MODY 5 or HNF1B-MODY, is a form of maturity onset diabetes of the young.
Shwachman–Diamond syndrome (SDS), or Shwachman–Bodian–Diamond syndrome, is a rare congenital disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, skeletal and cardiac abnormalities and short stature. After cystic fibrosis (CF), it is the second most common cause of exocrine pancreatic insufficiency in children. It is associated with the SBDS gene and has autosomal recessive inheritance.
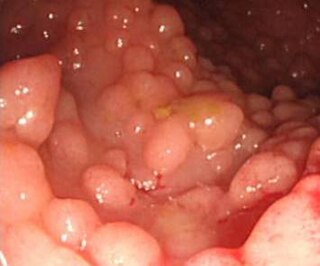
A hereditary cancer syndrome is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancer and may also cause early onset of these cancers. Hereditary cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.
The Emberger syndrome is a rare, autosomal dominant, genetic disorder caused by familial or sporadic inactivating mutations in one of the two parental GATA2 genes. The mutation results in a haploinsufficiency in the levels of the gene's product, the GATA2 transcription factor. This transcription factor is critical for the embryonic development, maintenance, and functionality of blood-forming, lympathic-forming, and other tissues. The syndrome includes as its primary symptoms: serious abnormalities of the blood such as the myelodysplastic syndrome and acute myeloid leukemia; lymphedema of the lower limbs, and sensorineural hearing loss. However, the anomalies caused by GATA2 mutations are highly variable with some individuals showing little or no such symptoms even in old age while others exhibit non-malignant types of hematological anomalies; lymphedema in areas besides the lower limbs, little or no hearing loss; or anomalies in other tissues. The syndrome may present with relatively benign signs and/or symptoms and then progress rapidly or slowly to the myelodysplastic syndrome and/or acute myeloid leukemia. Alternatively, it may present with one of the latter two life-threatening disorders.

