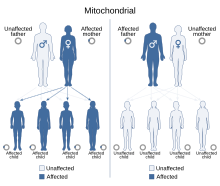
Mitochondrial DNA is the DNA located in mitochondria, cellular organelles within eukaryotic cells that convert chemical energy from food into a form that cells can use, such as adenosine triphosphate (ATP). Mitochondrial DNA is only a small portion of the DNA in a eukaryotic cell; most of the DNA can be found in the cell nucleus and, in plants and algae, also in plastids such as chloroplasts.

Mitochondrial disease is a group of disorders caused by mitochondrial dysfunction. Mitochondria are the organelles that generate energy for the cell and are found in every cell of the human body except red blood cells. They convert the energy of food molecules into the ATP that powers most cell functions.

Wolfram syndrome, also called DIDMOAD, is a rare autosomal-recessive genetic disorder that causes childhood-onset diabetes mellitus, optic atrophy, and deafness as well as various other possible disorders including neurodegeneration.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.

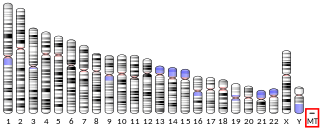
Human mitochondrial genetics is the study of the genetics of human mitochondrial DNA. The human mitochondrial genome is the entirety of hereditary information contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
Mitochondrially encoded 12S ribosomal RNA is the SSU rRNA of the mitochondrial ribosome. In humans, 12S is encoded by the MT-RNR1 gene and is 959 nucleotides long. MT-RNR1 is one of the 37 genes contained in animal mitochondria genomes. Their 2 rRNA, 22 tRNA and 13 mRNA genes are very useful in phylogenetic studies, in particular the 12S and 16S rRNAs. The 12S rRNA is the mitochondrial homologue of the prokaryotic 16S and eukaryotic nuclear 18S ribosomal RNAs. Mutations in the MT-RNR1 gene may be associated with hearing loss. The rRNA gene also encodes a peptide MOTS-c, also known as Mitochondrial-derived peptide MOTS-c or Mitochondrial open reading frame of the 12S rRNA-c.
MT-ND2 is a gene of the mitochondrial genome coding for the NADH dehydrogenase 2 (ND2) protein. The ND2 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of human MT-ND2 are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS), Leber's hereditary optic neuropathy (LHON) and increases in adult BMI.
MT-ATP6 is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 6' that encodes the ATP synthase Fo subunit 6. This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase. This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP. Mutations in the MT-ATP6 gene have been found in approximately 10 to 20 percent of people with Leigh syndrome.
Mitochondrially encoded tRNA leucine 1 (UUA/G) also known as MT-TL1 is a transfer RNA which in humans is encoded by the mitochondrial MT-TL1 gene.
Mitochondrially encoded tRNA histidine, also known as MT-TH, is a transfer RNA which, in humans, is encoded by the mitochondrial MT-TH gene.
MT-ND1 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 1 (ND1) protein. The ND1 protein is a subunit of NADH dehydrogenase, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of the human MT-ND1 gene are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS), Leber's hereditary optic neuropathy (LHON) and increases in adult BMI.
Mitochondrially encoded tRNA glutamic acid also known as MT-TE is a transfer RNA which in humans is encoded by the mitochondrial MT-TE gene. MT-TE is a small 69 nucleotide RNA that transfers the amino acid glutamic acid to a growing polypeptide chain at the ribosome site of protein synthesis during translation.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA asparagine also known as MT-TN is a transfer RNA which in humans is encoded by the mitochondrial MT-TN gene.
Mitochondrially encoded tRNA arginine also known as MT-TR is a transfer RNA which in humans is encoded by the mitochondrial MT-TR gene.

The mitochondrial ribosome, or mitoribosome, is a protein complex that is active in mitochondria and functions as a riboprotein for translating mitochondrial mRNAs encoded in mtDNA. The mitoribosome is attached to the inner mitochondrial membrane. Mitoribosomes, like cytoplasmic ribosomes, consist of two subunits — large (mt-LSU) and small (mt-SSU). Mitoribosomes consist of several specific proteins and fewer rRNAs. While mitochondrial rRNAs are encoded in the mitochondrial genome, the proteins that make up mitoribosomes are encoded in the nucleus and assembled by cytoplasmic ribosomes before being implanted into the mitochondria.