Mitochondrial disease is a group of disorders caused by mitochondrial dysfunction. Mitochondria are the organelles that generate energy for the cell and are found in every cell of the human body except red blood cells. They convert the energy of food molecules into the ATP that powers most cell functions.

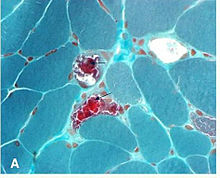
Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. On biopsy, the muscle tissue of patients with these diseases usually demonstrate "ragged red" muscle fibers. These ragged-red fibers contain mild accumulations of glycogen and neutral lipids, and may show an increased reactivity for succinate dehydrogenase and a decreased reactivity for cytochrome c oxidase. Inheritance was believed to be maternal. It is now known that certain nuclear DNA deletions can also cause mitochondrial myopathy such as the OPA1 gene deletion. There are several subcategories of mitochondrial myopathies.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.
Ocular myasthenia gravis (MG) is a disease of the neuromuscular junction resulting in hallmark variability in muscle weakness and fatigability. MG is an autoimmune disease where anomalous antibodies are produced against the naturally occurring acetylcholine receptors in voluntary muscles. MG may be limited to the muscles of the eye, leading to abrupt onset of weakness/fatigability of the eyelids or eye movement. MG may also involve other muscle groups.

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.

Ptosis, also known as blepharoptosis, is a drooping or falling of the upper eyelid. This condition is sometimes called "lazy eye," but that term normally refers to the condition amblyopia. If severe enough and left untreated, the drooping eyelid can cause other conditions, such as amblyopia or astigmatism, so it is especially important to treat the disorder in children before it can interfere with vision development.

Neuropathy, ataxia, and retinitis pigmentosa, also known as NARP syndrome, is a rare disease with mitochondrial inheritance that causes a variety of signs and symptoms chiefly affecting the nervous system Beginning in childhood or early adulthood, most people with NARP experience numbness, tingling, or pain in the arms and legs ; muscle weakness; and problems with balance and coordination (ataxia). Many affected individuals also have vision loss caused by changes in the light-sensitive tissue that lines the back of the eye. In some cases, the vision loss results from a condition called retinitis pigmentosa. This eye disease causes the light-sensing cells of the retina gradually to deteriorate.
Mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE) is a rare autosomal recessive mitochondrial disease. It has been previously referred to as polyneuropathy, ophthalmoplegia, leukoencephalopathy, and POLIP syndrome. The disease presents in childhood, but often goes unnoticed for decades. Unlike typical mitochondrial diseases caused by mitochondrial DNA (mtDNA) mutations, MNGIE is caused by mutations in the TYMP gene, which encodes the enzyme thymidine phosphorylase. Mutations in this gene result in impaired mitochondrial function, leading to intestinal symptoms as well as neuro-ophthalmologic abnormalities. A secondary form of MNGIE, called MNGIE without leukoencephalopathy, can be caused by mutations in the POLG gene.
Pearson syndrome is a mitochondrial disease characterized by sideroblastic anemia and exocrine pancreas dysfunction. Other clinical features are failure to thrive, pancreatic fibrosis with insulin-dependent diabetes and exocrine pancreatic deficiency, muscle and neurologic impairment, and, frequently, early death. It is usually fatal in infancy. The few patients who survive into adulthood often develop symptoms of Kearns–Sayre syndrome. It is caused by a deletion in mitochondrial DNA. Pearson syndrome is very rare, less than a hundred cases have been reported in medical literature worldwide.

DNA polymerase subunit gamma is an enzyme that in humans is encoded by the POLG gene. Mitochondrial DNA polymerase is heterotrimeric, consisting of a homodimer of accessory subunits plus a catalytic subunit. The protein encoded by this gene is the catalytic subunit of mitochondrial DNA polymerase. Defects in this gene are a cause of progressive external ophthalmoplegia with mitochondrial DNA deletions 1 (PEOA1), sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO), Alpers-Huttenlocher syndrome (AHS), and mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE).

Twinkle protein also known as twinkle mtDNA helicase is a mitochondrial protein that in humans is encoded by the TWNK gene located in the long arm of chromosome 10 (10q24.31).
DNA polymerase subunit gamma-2, mitochondrial is a protein that in humans is encoded by the POLG2 gene. The POLG2 gene encodes a 55 kDa accessory subunit protein that imparts high processivity and salt tolerance to the catalytic subunit of DNA polymerase gamma, encoded by the POLG gene. Mutations in this gene result in autosomal dominant progressive external ophthalmoplegia with mitochondrial DNA deletions.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA asparagine also known as MT-TN is a transfer RNA which in humans is encoded by the mitochondrial MT-TN gene.
Mitochondrially encoded tRNA tyrosine, also known as MT-TY, is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.
Multi/minicore myopathy is a congenital myopathy usually caused by mutations in either the SEPN1 and RYR1 genes. It is characterised the presence of multifocal, well-circumscribed areas with reduction of oxidative staining and low myofibrillar ATPase on muscle biopsy. It is also known as Minicore myopathy, Multicore myopathy, Multiminicore myopathy, Minicore myopathy with external ophthalmoplegia, Multicore myopathy with external ophthalmoplegia and Multiminicore disease with external ophthalmoplegia.

Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.
Sengers syndrome is a rare autosomal recessive condition characterised by congenital cataract, hypertrophic cardiomyopathy, muscle weakness and lactic acidosis after exercise. In some cases, they are inherited, in which case they would be called congenital. In addition, heart disease and muscle disease are prevalent, and life expectancy is short for many patients.
Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis, also known as SANDO syndrome, is a very rare genetic disorder which is characterized by ocular and nerve anomalies.