Angela Vincent is Emeritus professor at the University of Oxford and a Fellow of Somerville College, Oxford.

Lambert–Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder characterized by muscle weakness of the limbs.
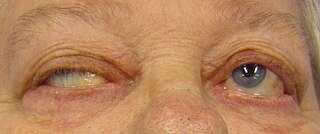
Myasthenia gravis (MG) is a long-term neuromuscular junction disease that leads to varying degrees of skeletal muscle weakness. The most commonly affected muscles are those of the eyes, face, and swallowing. It can result in double vision, drooping eyelids, and difficulties in talking and walking. Onset can be sudden. Those affected often have a large thymus or develop a thymoma.
Neuromyotonia (NMT) is a form of peripheral nerve hyperexcitability that causes spontaneous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. NMT along with Morvan's syndrome are the most severe types in the Peripheral Nerve Hyperexciteability spectrum. Example of two more common and less severe syndromes in the spectrum are Cramp Fasciculation Syndrome and Benign Fasciculation Syndrome. NMT can have both hereditary and acquired forms. The prevalence of NMT is unknown.

Acetylcholine (ACh) is an organic compound that functions in the brain and body of many types of animals as a neurotransmitter. Its name is derived from its chemical structure: it is an ester of acetic acid and choline. Parts in the body that use or are affected by acetylcholine are referred to as cholinergic.

An acetylcholine receptor is an integral membrane protein that responds to the binding of acetylcholine, a neurotransmitter.

An excitatory synapse is a synapse in which an action potential in a presynaptic neuron increases the probability of an action potential occurring in a postsynaptic cell. Neurons form networks through which nerve impulses travels, each neuron often making numerous connections with other cells of neurons. These electrical signals may be excitatory or inhibitory, and, if the total of excitatory influences exceeds that of the inhibitory influences, the neuron will generate a new action potential at its axon hillock, thus transmitting the information to yet another cell.
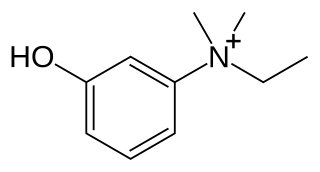
Edrophonium is a readily reversible acetylcholinesterase inhibitor. It prevents breakdown of the neurotransmitter acetylcholine and acts by competitively inhibiting the enzyme acetylcholinesterase, mainly at the neuromuscular junction. It is sold under the trade names Tensilon and Enlon.
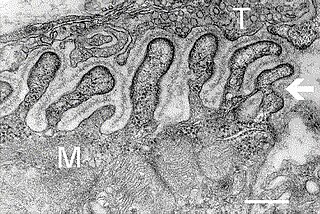
A neuromuscular junction is a chemical synapse between a motor neuron and a muscle fiber.
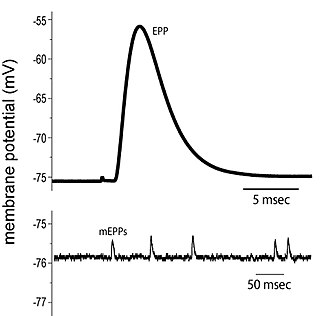
End plate potentials (EPPs) are the voltages which cause depolarization of skeletal muscle fibers caused by neurotransmitters binding to the postsynaptic membrane in the neuromuscular junction. They are called "end plates" because the postsynaptic terminals of muscle fibers have a large, saucer-like appearance. When an action potential reaches the axon terminal of a motor neuron, vesicles carrying neurotransmitters are exocytosed and the contents are released into the neuromuscular junction. These neurotransmitters bind to receptors on the postsynaptic membrane and lead to its depolarization. In the absence of an action potential, acetylcholine vesicles spontaneously leak into the neuromuscular junction and cause very small depolarizations in the postsynaptic membrane. This small response (~0.4mV) is called a miniature end plate potential (MEPP) and is generated by one acetylcholine-containing vesicle. It represents the smallest possible depolarization which can be induced in a muscle.
MuSK is a receptor tyrosine kinase required for the formation and maintenance of the neuromuscular junction. It is activated by a nerve-derived proteoglycan called agrin, which is similarly also required for neuromuscular junction formation.

A neuromuscular disease is any disease affecting the peripheral nervous system (PNS), the neuromuscular junctions, or skeletal muscles, all of which are components of the motor unit. Damage to any of these structures can cause muscle atrophy and weakness. Issues with sensation can also occur.

Agrin is a large proteoglycan whose best-characterised role is in the development of the neuromuscular junction during embryogenesis. Agrin is named based on its involvement in the aggregation of acetylcholine receptors during synaptogenesis. In humans, this protein is encoded by the AGRN gene.
Dok-7 is a non-catalytic cytoplasmic adaptor protein that is expressed specifically in muscle and is essential for the formation of neuromuscular synapses. Further, Dok-7 contains pleckstrin homology (PH) and phosphotyrosine-binding (PTB) domains that are critical for Dok-7 function. Finally, mutations in Dok-7 are commonly found in patients with limb-girdle congenital myasthenia.
Congenital myasthenic syndrome (CMS) is an inherited neuromuscular disorder caused by defects of several types at the neuromuscular junction. The effects of the disease are similar to Lambert-Eaton Syndrome and myasthenia gravis, the difference being that CMS is not an autoimmune disorder. There are only 600 known family cases of this disorder and it is estimated that its overall frequency in the human population is 1 in 200,000.

43 kDa receptor-associated protein of the synapse (rapsyn) is a protein that in humans is encoded by the RAPSN gene.

Acetylcholine receptor subunit epsilon is a protein that in humans is encoded by the CHRNE gene.
Acetylcholine receptor subunit beta is a protein that in humans is encoded by the CHRNB1 gene.
Repetitive nerve stimulation is a variant of the nerve conduction study where electrical stimulation is delivered to a motor nerve repeatedly several times per second. By observing the change in the muscle electrical response (CMAP) after several stimulations, a physician can assess for the presence of a neuromuscular junction disease, and differentiate between presynaptic and postsynaptic conditions. The test was first described by German neurologist Friedrich Jolly in 1895, and is also known as Jolly's test.

The active zone or synaptic active zone is a term first used by Couteaux and Pecot-Dechavassinein in 1970 to define the site of neurotransmitter release. Two neurons make near contact through structures called synapses allowing them to communicate with each other. As shown in the adjacent diagram, a synapse consists of the presynaptic bouton of one neuron which stores vesicles containing neurotransmitter, and a second, postsynaptic neuron which bears receptors for the neurotransmitter, together with a gap between the two called the synaptic cleft. When an action potential reaches the presynaptic bouton, the contents of the vesicles are released into the synaptic cleft and the released neurotransmitter travels across the cleft to the postsynaptic neuron and activates the receptors on the postsynaptic membrane.