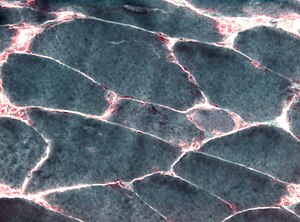
Inclusion body myositis (IBM) is the most common inflammatory muscle disease in older adults. The disease is characterized by slowly progressive weakness and wasting of both proximal muscles and distal muscles, most apparent in the finger flexors and knee extensors. IBM is often confused with an entirely different class of diseases, called hereditary inclusion body myopathies (hIBM). The "M" in hIBM is an abbreviation for "myopathy" while the "M" in IBM is for "myositis". In IBM, two processes appear to occur in the muscles in parallel, one autoimmune and the other degenerative. Inflammation is evident from the invasion of muscle fibers by immune cells. Degeneration is characterized by the appearance of holes, deposits of abnormal proteins, and filamentous inclusions in the muscle fibers. sIBM is a rare disease, with a prevalence ranging from 1 to 71 individuals per million.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. This results in muscular weakness. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere. Muscle cramps, stiffness, and spasm can also be associated with myopathy.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.

Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
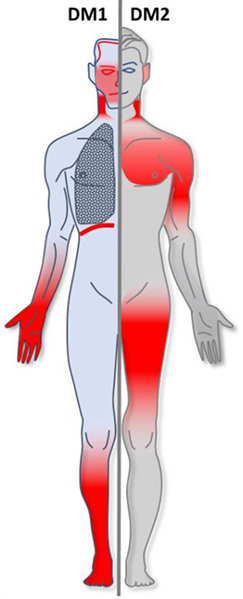
Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness. In DM, muscles are often unable to relax after contraction. Other manifestations may include cataracts, intellectual disability and heart conduction problems. In men, there may be early balding and an inability to father children. While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.
Congenital myopathy is a very broad term for any muscle disorder present at birth. This defect primarily affects skeletal muscle fibres and causes muscular weakness and/or hypotonia. Congenital myopathies account for one of the top neuromuscular disorders in the world today, comprising approximately 6 in 100,000 live births every year. As a whole, congenital myopathies can be broadly classified as follows:

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.

Central core disease (CCD), also known as central core myopathy, is an autosomal dominant inherited muscle disorder present from birth that negatively affects the skeletal muscles. It was first described by Shy and Magee in 1956. It is characterized by the appearance of the myofibril under the microscope.
Danon disease is a metabolic disorder. Danon disease is an X-linked lysosomal and glycogen storage disorder associated with hypertrophic cardiomyopathy, skeletal muscle weakness, and intellectual disability. It is inherited in an X-linked dominant pattern.

Neutral lipid storage disease is a congenital autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of leukocytes, muscle, liver, fibroblasts, and other tissues. It commonly occurs as one of two subtypes, cardiomyopathic neutral lipid storage disease (NLSD-M), or ichthyotic neutral lipid storage disease (NLSD-I) which is also known as Chanarin–Dorfman syndrome), which are characterized primarily by myopathy and ichthyosis, respectively. Normally, the ichthyosis that is present is typically non-bullous congenital ichthyosiform erythroderma which appears as white scaling.
Mitochondrially encoded tRNA glutamic acid also known as MT-TE is a transfer RNA which in humans is encoded by the mitochondrial MT-TE gene. MT-TE is a small 69 nucleotide RNA that transfers the amino acid glutamic acid to a growing polypeptide chain at the ribosome site of protein synthesis during translation.
Mitochondrially encoded tRNA phenylalanine also known as MT-TF is a transfer RNA which in humans is encoded by the mitochondrial MT-TF gene.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Desmin-related myofibrillar myopathy, is a subgroup of the myofibrillar myopathy diseases and is the result of a mutation in the gene that codes for desmin which prevents it from forming protein filaments, instead forming aggregates of desmin and other proteins throughout the cell.
Sporadic late-onset nemaline myopathy, or SLONM, is a very rare disease, one of the nemaline myopathies, causing loss of muscle bulk and weakness in the legs but sparing the cranial nerves, and beginning its clinical course after age 40. It was first identified in 1966 at the Mayo Clinic, by A.G. Engel, and that same year W.K. Engel and J.S. Resnick noted another case that they elaborated in 1975. The diagnosis of the disease rests on subacutely evolving weakness after age 40, normal to low CK level, a myopathic EMG with fibrillations, and often a monoclonal gammopathy. The diagnosis is confirmed by visualizing rods in cryosections on light and electron microscopy. The associated monoclonal gammopathy has an unfavorable prognosis.
Autophagic vacuolar myopathy (AVM) consists of multiple rare genetic disorders with common histological and pathological features on muscle biopsy. The features highlighted are vacuolar membranes of the autophagic vacuoles having sarcolemmal characteristics and an excess of autophagic vacuoles. There are currently five types of AVM identified. The signs and symptoms become more severe over the course of the disease. It begins with an inability to pick up small objects and progresses to difficulty in walking. The age of onset varies from early childhood to late adulthood, affecting people of all ages.