Diagnosis
| | This section is empty. You can help by adding to it. (July 2018) |
| Periodic Fever, Aphthous Stomatitis, Pharyngitis and Adenitis | |
|---|---|
| Other names | Periodic fever aphthous pharyngitis and cervical adenopathy (PFAPA) |
| Specialty | Pediatric, Rheumatology, Immunology |
| Symptoms | Fever recurring on a ~2-6 week cycle |
| Treatment | Tonsillectomy |
| Medication | Corticosteroids, Colchicine, Cimetidine |
Periodic fever, aphthous stomatitis, pharyngitis and adenitis syndrome is a medical condition, typically occurring in young children, in which high fever occurs periodically at intervals of about 3-5 weeks, frequently accompanied by aphthous-like ulcers, pharyngitis and/or cervical adenitis (cervical lymphadenopathy). The syndrome was described in 1987 and named two years later. [1] [2] [3]
The key symptoms of PFAPA are those in its name: periodic high fever at intervals of about 3–5 weeks, as well as aphthous ulcers, pharyngitis and/or adenitis. In between episodes, and even during the episodes, the children appear healthy. At least 6 months of episodes. Diagnosis requires recurrent negative throat cultures and that other causes (such as EBV, CMV, FMF) be excluded. [3]
The cause of PFAPA is unknown. [4] It is frequently discussed together with other periodic fever syndromes. [3]
Possible causes include primarily genetic factors or it may be due to an initial infection.
The condition appears to be the result of a disturbance of innate immunity. [5] The changes in the immune system are complex and include increased expression of complement related genes (C1QB, C2, SERPING1), interleukin-1-related genes (interleukin-1B, interleukin 1 RN, CASP1, interleukin 18 RAP) and interferon induced (AIM2, IP-10/CXCL10) genes. T cell associated genes (CD3, CD8B) are down regulated. Flares are accompanied by increased serum levels of activated T lymphocyte chemokines (IP-10/CXCL10, MIG/CXCL9), G-CSF and proinflammatory cytokines (interleukin 6, interleukin 18). Flares also manifest with a relative lymphopenia. Activated CD4(+)/CD25(+) T-lymphocyte counts correlated negatively with serum concentrations of IP-10/CXCL10, whereas CD4(+)/HLA-DR(+) T lymphocyte counts correlated positively with serum concentrations of the counterregulatory IL-1 receptor antagonist.[ citation needed ]
| | This section is empty. You can help by adding to it. (July 2018) |
PFAPA syndrome typically resolves spontaneously. Treatment options are used to lessen the severity of episodes. [6] These treatments are either medical or surgical:
One treatment often used is a dose of a corticosteroid at the beginning of each fever episode. [4] A single dose usually ends the fever within several hours. [4] However, in some children, they can cause the fever episodes to occur more frequently. [4] Interleukin-1 inhibition appears to be effective in treating this condition. [5]
There has been some evidence for the use of medications to reduce the frequency of flare-ups, including colchicine and cimetidine. [7]
Surgical removal of the tonsils appears to be beneficial compared to no surgery in symptom resolution and number of future episodes. [6] The evidence to support surgery is; however, of moderate quality. [6]
Children with PFAPA have an impaired quality of life, which may be treated via individual counseling. [8]
According to present research, PFAPA does not lead to other diseases and spontaneously resolves as the child gets older, with no long term physical effects. [2] [9] [10] However, PFAPA has been found in adults and may not spontaneously resolve. [11] [ non-primary source needed ] Children with PFAPA experience lower physical, emotional, and psychosocial functioning. [8] Their performance in school is also substantially impacted. [8]
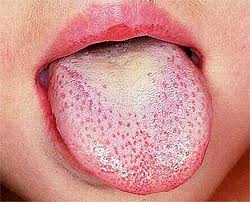
Scarlet fever, also known as scarlatina, is an infectious disease caused by Streptococcus pyogenes, a Group A streptococcus (GAS). The infection is a type of Group A streptococcal infection. It most commonly affects children between five and 15 years of age. The signs and symptoms include a sore throat, fever, headache, swollen lymph nodes, and a characteristic rash. The face is flushed and the rash is red and blanching. It typically feels like sandpaper and the tongue may be red and bumpy. The rash occurs as a result of capillary damage by exotoxins produced by S.pyogenes. On darker-pigmented skin the rash may be hard to discern.

Streptococcal pharyngitis, also known as streptococcal sore throat, is pharyngitis caused by Streptococcus pyogenes, a gram-positive, group A streptococcus. Common symptoms include fever, sore throat, red tonsils, and enlarged lymph nodes in the front of the neck. A headache and nausea or vomiting may also occur. Some develop a sandpaper-like rash which is known as scarlet fever. Symptoms typically begin one to three days after exposure and last seven to ten days.
Barth syndrome (BTHS) is a rare but serious X-linked genetic disorder, caused by changes in phospholipid structure and metabolism. It may affect multiple body systems, and is potentially fatal. The syndrome is diagnosed almost exclusively in males.

A mouth ulcer (aphtha) is an ulcer that occurs on the mucous membrane of the oral cavity. Mouth ulcers are very common, occurring in association with many diseases and by many different mechanisms, but usually there is no serious underlying cause. Rarely, a mouth ulcer that does not heal may be a sign of oral cancer. These ulcers may form individually or multiple ulcers may appear at once. Once formed, an ulcer may be maintained by inflammation and/or secondary infection.

Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.

Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence is often normal. Complications of NS can include leukemia.
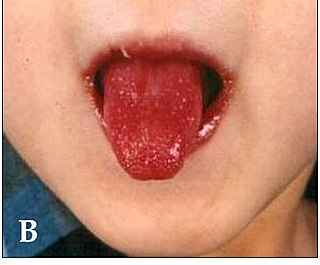
Kawasaki disease is a syndrome of unknown cause that results in a fever and mainly affects children under 5 years of age. It is a form of vasculitis, where medium-sized blood vessels become inflamed throughout the body. The fever typically lasts for more than five days and is not affected by usual medications. Other common symptoms include large lymph nodes in the neck, a rash in the genital area, lips, palms, or soles of the feet, and red eyes. Within three weeks of the onset, the skin from the hands and feet may peel, after which recovery typically occurs. The disease is the leading cause of acquired heart disease in children in developed countries, which include the formation of coronary artery aneurysms and myocarditis.

Aphthous stomatitis, or recurrent aphthous stomatitis (RAS), is a common condition characterized by the repeated formation of benign and non-contagious mouth ulcers (aphthae) in otherwise healthy individuals. The informal term canker sore is also used, mainly in North America, although it may also refer to other types of mouth ulcers. The cause is not completely understood but involves a T cell-mediated immune response triggered by a variety of factors which may include nutritional deficiencies, local trauma, stress, hormonal influences, allergies, genetic predisposition, certain foods, dehydration, some food additives, or some hygienic chemical additives like SDS.

Stomatitis is inflammation of the mouth and lips. It refers to any inflammatory process affecting the mucous membranes of the mouth and lips, with or without oral ulceration.
Cogan syndrome is a rare disorder characterized by recurrent inflammation of the front of the eye and often fever, fatigue, and weight loss, episodes of vertigo (dizziness), tinnitus and hearing loss. It can lead to deafness or blindness if untreated. The classic form of the disease was first described by D. G. Cogan in 1945.
Hyperimmunoglobulinemia E syndrome (HIES), of which the autosomal dominant form is called Job's syndrome or Buckley syndrome, is a heterogeneous group of immune disorders. Job's is also very rare at about 300 cases currently in the literature.

Muckle–Wells syndrome (MWS) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives, and can lead to amyloidosis. Individuals with MWS often have episodic fever, chills, and joint pain. As a result, MWS is considered a type of periodic fever syndrome. MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin. MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease—in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS).
Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.

Mevalonate kinase is an enzyme that in humans is encoded by the MVK gene. Mevalonate kinases are found in a wide variety of organisms from bacteria to mammals. This enzyme catalyzes the following reaction:

Spondylocostal dysostosis, also known as Jarcho-Levin syndrome (JLS), is a rare, heritable axial skeleton growth disorder. It is characterized by widespread and sometimes severe malformations of the vertebral column and ribs, shortened thorax, and moderate to severe scoliosis and kyphosis. Individuals with Jarcho-Levin typically appear to have a short trunk and neck, with arms appearing relatively long in comparison, and a slightly protuberant abdomen. Severely affected individuals may have life-threatening pulmonary complications due to deformities of the thorax. The syndrome was first described by Saul Jarcho and Paul M. Levin at Johns Hopkins University in 1938.
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare condition (1:1,000,000), in which the bones have lesions, inflammation, and pain. It is called multifocal because it can appear in different parts of the body, primarily bones, and osteomyelitis because it is very similar to that disease, although CRMO appears to be without any infection.

The Interleukin-2 receptor alpha chain is a protein involved in the assembly of the high-affinity Interleukin-2 receptor, consisting of alpha (IL2RA), beta (IL2RB) and the common gamma chain (IL2RG). As the name indicates, this receptor interacts with Interleukin-2, a pleiotropic cytokine which plays an important role in immune homeostasis.
Michael E. Pichichero is an American physician who is the Director of the Rochester General Hospital Research Institute, a Research Professor at Rochester Institute of Technology and a clinical professor in the Department of Pediatrics at the University of Rochester Medical Center. He is the author of a number of scientific studies regarding the safety of thimerosal as a preservative in vaccines.
Ann Stone Minot was an American biochemist and physiologist.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by dysfunction of the innate immune system. They are characterized by periodic or chronic systemic inflammation, usually without the involvement of adaptive immunity.