
H2 antagonists, sometimes referred to as H2RAs and also called H2 blockers, are a class of medications that block the action of histamine at the histamine H2 receptors of the parietal cells in the stomach. This decreases the production of stomach acid. H2 antagonists can be used in the treatment of dyspepsia, peptic ulcers and gastroesophageal reflux disease. They have been surpassed by proton pump inhibitors (PPIs). The PPI omeprazole was found to be more effective at both healing and alleviating symptoms of ulcers and reflux oesophagitis than the H2 blockers ranitidine and cimetidine.
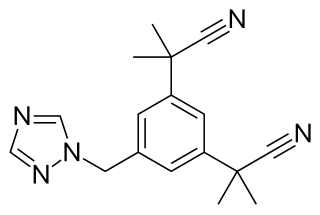
Anastrozole, sold under the brand name Arimidex among others, is an antiestrogenic medication used in addition to other treatments for breast cancer. Specifically it is used for hormone receptor-positive breast cancer. It has also been used to prevent breast cancer in those at high risk. It is taken by mouth.

Famotidine, sold under the brand name Pepcid among others, is a histamine H2 receptor antagonist medication that decreases stomach acid production. It is used to treat peptic ulcer disease, gastroesophageal reflux disease, and Zollinger-Ellison syndrome. It is taken by mouth or by injection into a vein. It begins working within an hour.
Cytochrome P450 3A4 is an important enzyme in the body, mainly found in the liver and in the intestine, which in humans is encoded by CYP3A4 gene. It oxidizes small foreign organic molecules (xenobiotics), such as toxins or drugs, so that they can be removed from the body. It is highly homologous to CYP3A5, another important CYP3A enzyme.

Dextrorphan (DXO) is a psychoactive drug of the morphinan class which acts as an antitussive or cough suppressant and in high doses a dissociative hallucinogen. It is the dextrorotatory enantiomer of racemorphan; the levorotatory enantiomer is levorphanol. Dextrorphan is produced by O-demethylation of dextromethorphan by CYP2D6. Dextrorphan is an NMDA antagonist and contributes to the psychoactive effects of dextromethorphan.

Bicalutamide, sold under the brand name Casodex among others, is an antiandrogen medication that is primarily used to treat prostate cancer. It is typically used together with a gonadotropin-releasing hormone (GnRH) analogue or surgical removal of the testicles to treat metastatic prostate cancer (mPC). To a lesser extent, it is used at high doses for locally advanced prostate cancer (LAPC) as a monotherapy without castration. Bicalutamide was also previously used as monotherapy to treat localized prostate cancer (LPC), but authorization for this use was withdrawn following unfavorable trial findings. Besides prostate cancer, bicalutamide is limitedly used in the treatment of excessive hair growth and scalp hair loss in women, as a puberty blocker and component of feminizing hormone therapy for transgender girls and women, to treat gonadotropin-independent early puberty in boys, and to prevent overly long-lasting erections in men. It is taken by mouth.

Tamoxifen, sold under the brand name Nolvadex among others, is a selective estrogen receptor modulator used to prevent breast cancer in women and men. It is also being studied for other types of cancer. It has been used for Albright syndrome. Tamoxifen is typically taken daily by mouth for five years for breast cancer.

Nortriptyline, sold under the brand name Pamelor, among others, is a medication used to treat depression. This medicine is also sometimes used for neuropathic pain, attention deficit hyperactivity disorder (ADHD), smoking cessation and anxiety. As with many antidepressants, its use for young people with depression and other psychiatric disorders may be limited due to increased suicidality in the 18–24 population initiating treatment. Nortriptyline is a less preferred treatment for ADHD and stopping smoking. It is taken by mouth.

Doxepin is a medication belonging to the tricyclic antidepressant (TCA) class of drugs used to treat major depressive disorder, anxiety disorders, chronic hives, and insomnia. For hives it is a less preferred alternative to antihistamines. It has a mild to moderate benefit for sleeping problems. It is used as a cream for itchiness due to atopic dermatitis or lichen simplex chronicus.

Metiamide is a histamine H2 receptor antagonist developed from another H2 antagonist, burimamide. It was an intermediate compound in the development of the successful anti-ulcer drug cimetidine (Tagamet).

Antihistamines are drugs which treat allergic rhinitis, common cold, influenza, and other allergies. Typically, people take antihistamines as an inexpensive, generic drug that can be bought without a prescription and provides relief from nasal congestion, sneezing, or hives caused by pollen, dust mites, or animal allergy with few side effects. Antihistamines are usually for short-term treatment. Chronic allergies increase the risk of health problems which antihistamines might not treat, including asthma, sinusitis, and lower respiratory tract infection. Consultation of a medical professional is recommended for those who intend to take antihistamines for longer-term use.
Antiestrogens, also known as estrogen antagonists or estrogen blockers, are a class of drugs which prevent estrogens like estradiol from mediating their biological effects in the body. They act by blocking the estrogen receptor (ER) and/or inhibiting or suppressing estrogen production. Antiestrogens are one of three types of sex hormone antagonists, the others being antiandrogens and antiprogestogens. Antiestrogens are commonly used to stop steroid hormones, estrogen, from binding to the estrogen receptors leading to the decrease of estrogen levels. Decreased levels of estrogen can lead to complications in sexual development. Antiandrogens are sex hormone antagonists which are able to lower the production and the effects that testosterone can have on female bodies.
Feminizing hormone therapy, also known as transfeminine hormone therapy, is hormone therapy and sex reassignment therapy to change the secondary sex characteristics of transgender people from masculine or androgynous to feminine. It is a common type of transgender hormone therapy and is used to treat transgender women and non-binary transfeminine individuals. Some, in particular intersex people, but also some non-transgender people, take this form of therapy according to their personal needs and preferences.

Afimoxifene, also known as 4-hydroxytamoxifen (4-OHT) and by its tentative brand name TamoGel, is a selective estrogen receptor modulator (SERM) of the triphenylethylene group and an active metabolite of tamoxifen. The drug is under development under the tentative brand name TamoGel as a topical gel for the treatment of hyperplasia of the breast. It has completed a phase II clinical trial for cyclical mastalgia, but further studies are required before afimoxifene can be approved for this indication and marketed.

Benorterone, also known by its developmental code name SKF-7690 and as 17α-methyl-B-nortestosterone, is a steroidal antiandrogen which was studied for potential medical use but was never marketed. It was the first known antiandrogen to be studied in humans. It is taken by mouth or by application to skin.
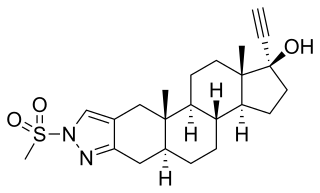
Zanoterone, also known as (5α,17α)-1'-(methylsulfonyl)-1'-H-pregn-20-yno[3,2-c]pyrazol-17-ol, is a steroidal antiandrogen which was never marketed. It was investigated for the treatment of benign prostatic hyperplasia (BPH) but failed to demonstrate sufficient efficacy in phase II clinical trials, and also showed an unacceptable incidence rate and severity of side effects. As such, it was not further developed.
A nonsteroidal antiandrogen (NSAA) is an antiandrogen with a nonsteroidal chemical structure. They are typically selective and full or silent antagonists of the androgen receptor (AR) and act by directly blocking the effects of androgens like testosterone and dihydrotestosterone (DHT). NSAAs are used in the treatment of androgen-dependent conditions in men and women. They are the converse of steroidal antiandrogens (SAAs), which are antiandrogens that are steroids and are structurally related to testosterone.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

Bifluranol is a synthetic nonsteroidal estrogen of the stilbestrol group related to diethylstilbestrol that has been used as an antiandrogen in the United Kingdom in the treatment of benign prostatic hyperplasia. The drug is described as a weak estrogen, and possesses about one-eighth the potency of diethylstilbestrol.

The pharmacodynamics of spironolactone, an antimineralocorticoid and antiandrogen medication, concern its mechanisms of action, including its biological targets and activities, as well as its physiological effects. The pharmacodynamics of spironolactone are characterized by high antimineralocorticoid activity, moderate antiandrogenic activity, and weak steroidogenesis inhibition. In addition, spironolactone has sometimes been found to increase estradiol and cortisol levels and hence could have slight indirect estrogenic and glucocorticoid effects. The medication has also been found to interact very weakly with the estrogen and progesterone receptors, and to act as an agonist of the pregnane X receptor. Likely due to increased activation of the estrogen and/or progesterone receptors, spironolactone has very weak but significant antigonadotropic effects.

